A hatásmechanizmusok gyógyászati \u200b\u200banyagok - ezek az anyagok okozhatják a farmakológiai hatásokat. A kábítószerek fő hatásmechanizmusai a következők:
Fizikai.
A közvetlen kémiai kölcsönhatás mechanizmusa.
Membrán (fizikai-kémiai).
Enzimatikus (biokémiai).
Receptor.
A működés fizikai mechanizmusa. A gyógyszer hatása összekapcsolódik annak fizikai tulajdonságaival. Például az aktív szént speciálisan feldolgozzák, ezért magas felületi aktivitással bírnak. Ez lehetővé teszi számára, hogy felszívja a gázokat, alkaloidokat, toxinokat stb.
Közvetlen kémiai kölcsönhatás. Ez a gyógyszerek meglehetősen ritka hatásmechanizmusa, amelynek lényege, hogy a gyógyszerek közvetlenül kölcsönhatásba lépnek a testben levő molekulákkal vagy ionokkal. Ilyen hatásmechanizmust tartalmaz például az antidotumok csoportjába tartozó unitiol gyógyszer. Tiolmérgekkel - beleértve a nehézfémek sóit - történő mérgezés esetén az unitiol közvetlen kémiai reakcióba lép velük, és nem toxikus komplexek képződését eredményezheti, amelyek kiválasztódnak a vizelettel. Tehát az antacidok is hatnak, amelyek közvetlen kémiai kölcsönhatásba lépnek a sósavval, csökkentve a gyomornedv savasságát.
hártyás (Fizikai-kémiai) a mechanizmus. Ehhez kapcsolódik a gyógyszerek ionáramokra gyakorolt \u200b\u200bhatása (Na +, K +, Cl - és mások), amelyek meghatározzák a transzmembrán elektromos potenciált. E mechanizmus szerint érzéstelenítők, antiaritmiás szerek, helyi érzéstelenítők stb.
enzimatikus (Biochemistry) a mechanizmus. Ezt a mechanizmust az határozza meg, hogy egyes gyógyszerek képesek-e aktiválni vagy gátolni az enzimeket. Az ilyen hatásmechanizmussal rendelkező gyógyszerek arzenálja nagyon széles. Például antikolineszteráz gyógyszerek, monoamin-oxidáz inhibitorok, protonpumpa-blokkolók stb.
Receptor mechanizmus. Az emberi testben vannak nagyon specifikus biológiailag aktív anyagok (mediátorok), amelyek kölcsönhatásba lépnek a receptorokkal, és megváltoztatják a test különféle szerveinek vagy szöveteinek működését.
A receptorok makromolekuláris struktúrák, amelyek szelektív érzékenységet mutatnak bizonyos kémiai vegyületekkel szemben. A gyógyszerek és a receptorok kölcsönhatása során biokémiai és élettani változások lépnek fel a testben, amelyeket egy vagy másik klinikai hatás kísér.
Közvetítőket és gyógyszereket hívnak, amelyek aktiválják a receptorokat és biológiai hatást keltenek agonisták. Azokat a gyógyászati \u200b\u200banyagokat, amelyek kötődnek a receptorokhoz, de nem idézik elő aktiválásukat és biológiai hatásaikat, csökkentik vagy kiküszöbölik az agonisták hatásait, nevezzük antagonisták. Kiosztani is antagonista agonisták - olyan anyagok, amelyek ugyanazon receptorok altípusain eltérően hatnak: stimulálnak egyes receptor altípusokat, és blokkolnak másokat. Például a narkotikus fájdalomcsillapító nalbufin stimulálja az opioid kappa receptorokat (ennélfogva csökkenti a fájdalomérzékenységet) és blokkolja az opioid mu receptorokat (ennélfogva kevésbé veszélyes a drogfüggőség szempontjából).
Az anyagoknak a receptorokhoz való kötődésének képességére az „affinitás” kifejezés utal. Ugyanazon receptorokhoz viszonyítva a különböző anyagok affinitása eltérő lehet.
A következő típusú receptorokat különböztetjük meg:
Plazmamembrán receptorok:
csatorna típusa: N-típusú kolinerg receptorok, izom-típusú H-kolinerg receptorok, GABA-receptorok;
g-protein receptorok: α- és β-adrenerg receptorok, M3-kolinoreceptorok;
integráló típusú receptorok: NO receptor.
Citoszolikus.
Mitokondriális.
A farmakodinamika egy szakasz általános farmakológiaa drogok testre gyakorolt \u200b\u200bhatásainak tanulmányozása. Nevezetesen, a farmakodinamikai vizsgálatok:
- a drogok hatásmechanizmusai;
- vég farmakológiai hatások;
- a kábítószerek hatásának különféle feltételektől való függése;
- a gyógyszerek hatása az ismételt beadásra;
- a kábítószerek kombinált hatása;
- gyógyszer-összeférhetetlenség;
- gyógyszerek mellékhatásai.
A hatásmechanizmusok gyógyszerek
A gyógyszerek hatásmechanizmusa az, ahogyan az anyagok farmakológiai hatásokat idéznek elő. A kábítószerek fő hatásmechanizmusai a következők:
- Fizikai.
- A közvetlen kémiai kölcsönhatás mechanizmusa.
- Membrán (fizikai-kémiai).
- Enzimatikus (biokémiai).
- Receptor.
A működés fizikai mechanizmusa. A gyógyszer hatása összekapcsolódik annak fizikai tulajdonságaival. Például az aktív szént speciálisan feldolgozzák, ezért magas felületi aktivitással bírnak. Ez lehetővé teszi számára, hogy felszívja a gázokat, alkaloidokat, toxinokat stb.
Közvetlen kémiai kölcsönhatás. Ez a gyógyszerek meglehetősen ritka hatásmechanizmusa, amelynek lényege, hogy a gyógyszerek közvetlenül kölcsönhatásba lépnek a testben levő molekulákkal vagy ionokkal. Ilyen hatásmechanizmust tartalmaz például az antidotumok csoportjába tartozó unitiol gyógyszer. Tiolmérgekkel - beleértve a nehézfémek sóit - történő mérgezés esetén az unitiol közvetlen kémiai reakcióba lép velük, és nem toxikus komplexek képződését eredményezheti, amelyek kiválasztódnak a vizelettel. Tehát az antacidok is hatnak, amelyek közvetlen kémiai kölcsönhatásba lépnek a sósavval, csökkentve a gyomornedv savasságát.
hártyás (Fizikai-kémiai) a mechanizmus. Ehhez kapcsolódik a gyógyszerek ionáramokra gyakorolt \u200b\u200bhatása (Na +, K +, Cl - és mások), amelyek meghatározzák a transzmembrán elektromos potenciált. E mechanizmus szerint érzéstelenítők, antiaritmiás szerek, helyi érzéstelenítők stb.
enzimatikus (Biochemistry) a mechanizmus. Ezt a mechanizmust az határozza meg, hogy egyes gyógyszerek képesek-e aktiválni vagy gátolni az enzimeket. Az ilyen hatásmechanizmussal rendelkező gyógyszerek arzenálja nagyon széles. Például antikolineszteráz gyógyszerek, monoamin-oxidáz inhibitorok, protonpumpa-blokkolók stb.
Receptor mechanizmus. Az emberi testben vannak nagyon specifikus biológiailag aktív anyagok (mediátorok), amelyek kölcsönhatásba lépnek a receptorokkal, és megváltoztatják a test különféle szerveinek vagy szöveteinek működését.
A receptorok makromolekuláris struktúrák, amelyek szelektív érzékenységet mutatnak bizonyos kémiai vegyületekkel szemben. A gyógyszerek és a receptorok kölcsönhatása során biokémiai és élettani változások lépnek fel a testben, amelyeket egy vagy másik klinikai hatás kísér.
Közvetítőket és gyógyszereket hívnak, amelyek aktiválják a receptorokat és biológiai hatást keltenek agonisták. Azokat a gyógyászati \u200b\u200banyagokat, amelyek kötődnek a receptorokhoz, de nem idézik elő aktiválásukat és biológiai hatásaikat, csökkentik vagy kiküszöbölik az agonisták hatásait, nevezzük antagonisták. Kiosztani is antagonista agonisták - olyan anyagok, amelyek ugyanazon receptorok altípusain eltérően hatnak: stimulálnak egyes receptor altípusokat, és blokkolnak másokat. Például a narkotikus fájdalomcsillapító nalbufin stimulálja az opioid kappa receptorokat (ennélfogva csökkenti a fájdalomérzékenységet) és blokkolja az opioid mu receptorokat (ennélfogva kevésbé veszélyes a drogfüggőség szempontjából).
Az anyagoknak a receptorokhoz való kötődésének képességére az „affinitás” kifejezés utal. Ugyanazon receptorokhoz viszonyítva a különböző anyagok affinitása eltérő lehet.
A következő típusú receptorokat különböztetjük meg:
- Plazmamembrán receptorok:
- csatorna típusa: N-típusú kolinerg receptorok, izom-típusú H-kolinerg receptorok, GABA-receptorok;
- g-protein receptorok: α- és β-adrenerg receptorok, M3-kolinoreceptorok;
- integráló típusú receptorok: NO receptor.
- Citoszolikus.
- Mitokondriális.
- Nukleáris.
Plazmamembrán receptorok.
Csatorna típusú receptorok
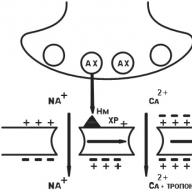
Az ideg típusú N n-kolinerg receptor (Központi idegrendszer, autonóm ganglionok, sinocarotid zóna, kromaffin mellékvesék szövete). Miután az acetilkolint (AX) Hn-kolinerg receptorokkal kötötték, a Na + csatornák kinyílnak, és a Na rohan a sejtbe, pozitív töltés mellett. A posztszinaptikus membrán depolarizált. Van egy akciópotenciál, amely eltolódik a neuron membránja mentén, nyitva az elektromosan függő Na + csatornákat. Idegimpulzus keletkezik a posztganglionikus rostban (6. ábra).
Ábra. 6. N n-kolinerg receptor
N m - izom típusú kolinerg receptor (vázizomsejt-membránok). A kezdeti folyamatok hasonlóak, de az elektromosan függő Ca ++ csatornák nyitva vannak. A Ca ++ ionok belépnek az izomrostokba, a Ca ++ felszabadul a sarkoplazmás retikulumból. A Ca ++ szintje emelkedik, ami izom-összehúzódást vált ki (7. ábra).

Ábra. 7. N m-kolinerg receptor
GABA receptorok. Ezek a γ-amino-vajsav (GABA) receptorai. A GABA kölcsönhatásba lép a GABA receptorokkal, amelyek szerkezetében klorid csatornák vannak. A receptor stimuláció eredményeként a csatornák nyitva vannak és a klórionok (Cl -) szabadon lépnek be a sejtbe. A klórionok koncentrációjának növekedése a sejtben a membrán hiperpolarizációjához és az idegsejtek aktivitásának csökkenéséhez vezet. Nehezebb ilyen sejt gerjesztése (8. ábra).

Ábra. 8. GABA receptor:
GABA-R - GABA-receptor, BD-R - benzodiazepin-receptor, BR - barbiturát-receptor
A receptorai G fehérje
A G-fehérjék, azaz a GTP-kötő (guanozin-trifoszfát-kötő) fehérjék a sejtmembránban lokalizálódnak, és α-, β- és γ-alegységekből állnak. Ezek (G-fehérjék) szabályozzák a specifikus effektorok (azonnali hírvivők, másodlagos közvetítők) aktivitását. Ezek a hírvivők lehetnek enzma (adenilát-cikláz, foszfolipáz); csatornák a káliumhoz, kalciumhoz, nátriumhoz; néhány transzportfehérje. Minden sejtben sok G-fehérje lehet, mindegyik szabályozza a különféle hírvivők aktivitását, miközben megváltoztatja a sejt funkcióját.
M3-kolinerg receptor (simaizommembránok (MMC) és exokrin mirigysejtek). Az acetilkolin stimulálja a G-proteinhez kötött M 3-XR-t. A foszfolipáz-C (FLS) aktiválódik, amely katalizálja a PIDP (foszfatidil-inozit-difoszfát) hasítását ITP-re (inozitol-trifoszfát) és DAG-ra (diacil-glicerin). Az MMC citoplazmájába belépő ITF felszabadítja a Ca ++ -ot a kagylókból .

Ábra. 9. M3-kolinerg receptor
A Ca ++ kötődik a kalodulinhoz, aktiválja a miozin-kinázt (MK), amely katalizálja a miozin könnyű láncának foszforilációját, ami a sejtek összehúzódásához vezet (9. ábra). Hasonlóképpen, egy impulzus továbbítódik a szekréciós mirigyek szinapszisán.
A noorepinefrin stimulálja α 1 -adrenoreceptorA következő eseménylánc elindításával:
Norepinefrin (HA) → α 1 -adrenoreceptor → a G s-protein α-alegységének aktiválása → a PLL aktiválása → FIDF hasítás → az ITF koncentráció növekedése → a Ca 2+ koncentráció növekedése a sejtben → Ca 2+ kötődik a kalodulinhoz → a miozin-kináz aktiválódik → könnyű láncok foszforilálódnak → könnyű láncok miozin → miozin kölcsönhatásba lép aktinnal → csökken az MMC csökkenése (10. ábra).

Ábra. 10. α 1 -adrenoreceptor
b 1 receptor(11. ábra). Norepinefrin → aktiválja a b 1-AP-t → a G-protein α-alegységének aktiválását → az AC aktiválását → a cAMP-termelés növekedését az ATP-ből → a cAMP-koncentráció növekedését a cardiomyocytában → protein-kinázok aktiválását → a kalciumcsatorna-fehérjék foszforilációját → a Ca 2+ bejutásának növekedését a csatornákon keresztül és növeli a Ca-koncentrációt 2+ a sejtben → a szív összehúzódásainak fokozódása.

Ábra. 11. b 1 receptor
b 2 receptor(12. ábra). BE → b 2 -AP → a G-protein α-alegységének aktiválása → AC aktiválása → fokozott cAMP képződés → stimulált protein-kináz → a miozin-kináz foszforilációját katalizáló kináz hasad, míg az utóbbi aktivitása elveszik → a miozin-foszforiláció nem fordul elő → MMC relaxáció.
A HA idegvégződésekből történő felszabadulásának szabályozását maga a neurotranszmitter végzi az α 2 -AP preszinaptikus membrán gerjesztésekor. A HA kibocsátása csökken.

Ábra. 12. b2 receptor
Integráló típusú receptorok
Ezek olyan receptorok, amelyek olyan fehérjék, amelyek áthatolnak a membránon. Ebben az esetben a fehérje külső része receptor szerepet játszik, míg a belső része katalitikus szerepet játszik (13. ábra).

Ábra. 13. Integráló típusú receptor
Citoszol receptorok
Fiziológiai körülmények között az ilyen receptorok szteroidhormonok (nemi hormonok, glükokortikoidok) megkötését szolgálják. Ezek az anyagok belépnek a sejtekbe, és ott kötődnek citoszol receptorokhoz. Ez a komplex behatol a magba, és megváltoztatja a genom működését. Ennek eredményeként megváltozik a fehérje szintézise a sejtben (14. ábra).
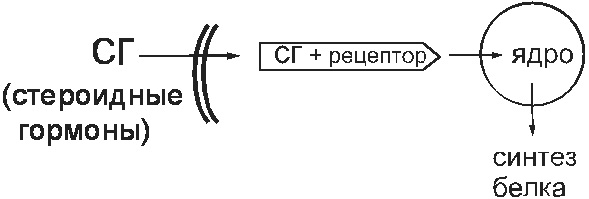
Ábra. 14. Citoszol receptor
Mitokondriális receptorok
A mitokondriumokban vannak olyan receptorok is, amelyekkel a gyógyhatású anyagok kölcsönhatásba lépnek, például a trijódtironin-hidroklorid, amelyek a természetes T 3 hormon analógjai. Ennek a kölcsönhatásnak az eredményeként az ATP szintézise növekszik.
Nukleáris receptorok
A T 3 behatol a magba, és kölcsönhatásba lép az ilyen típusú receptorokkal. Ennek eredményeként megváltozik a genom működése és új fehérjék szintetizálódnak.
Végső farmakológiai hatások (Vershinin szerint)
A gyógyszerek sokasága ellenére az általuk okozott változások a testben azonos típusúak (15. ábra). Bármely gyógyszernek a szervekre gyakorolt \u200b\u200bhatása öt fő farmakológiai hatásra csökkenthető (N.V. Vershinin szerint):
- kibékítés - a fokozott szervfunkció normalizálása (nyugtatók használata).
- elnyomás - a test funkciójának normája alá esik (drogok használata érzéstelenítéshez).
- bénulás - a csökkent szervfunkció leállítása (légzésdepresszió narkotikus fájdalomcsillapítók túladagolása esetén).
- Nyugtató - a csökkent funkció normalizálása (β 1 -adrenomimetikumok használata).
- ingerlés - A szerv funkciójának a normát meghaladó fokozása (diuretikumok használata mérgezés esetén, köpködő gyógyszerek).
Ábra. 15. Végleges farmakológiai hatások

A drogok hatásmódjai
- A lényeg másodlagos.
A legfontosabb egy hatás az, amely a gyógyszer terápiás vagy profilaktikus beadásának alapját képezi. oldalsó - nemkívánatos, veszélyes a gyógyszerek beteg általi hatására.
- Megfordítható, visszafordíthatatlan.
A testben lévő gyógyászati \u200b\u200banyagok kölcsönhatásba lépnek azokkal a sejtekkel, amelyeknek biológiai szubsztrátja képes reagálni az anyaggal. Ez a kölcsönhatás a gyógyszer kémiai szerkezetétől függ. A gyógyszer-anyag megfelelő szubsztrátumhoz kötődik megfordítható, ha (szubsztrát és gyógyszer) egy ideig kötődnek egymáshoz.
Néhány esetben a terápiás cél megköveteli visszafordíthatatlan egy szerkezet kikapcsolása a funkciójából. Ez vonatkozik például a legtöbb antimikrobiális, daganatellenes szerekre, amelyek képesek erős (kovalens) kötéseket képezni a DNS-hélix sejtek ("térhálósítás") elemekkel vagy a bakteriális enzimekkel, amelynek eredményeként a sejtek elveszítik reprodukciós képességüket.
- Közvetlen, közvetett (közvetett).
közvetlen a hatás azt jelenti, hogy a terápiás hatás a gyógyszer közvetlen kölcsönhatásából adódik a beteg szerv biosubsztrátjával, és közvetlenül bizonyos eltolódásokhoz vezet. Ha ennek eredményeként a szerv (rendszer) funkciója másodszor megváltozik közvetlen befolyás kábítószer egy másik szervre, egy másik rendszerre, ezt a hatást indirektnek (közvetettnek) hívják. A szívglikozidok javítják a szívizom kontraktilitását ( közvetlen fellépés), és ennek eredményeként javítja a test vérkeringését, amelyet a diurézis javulása (közvetett hatás) kísér.
A közvetett cselekvés különleges esete: reflex akció. Például a vazodilatáció és a trofikus szövetek javulása a bőr szenzoros idegeinek végeinek irritációja eredményeként.
- Szelektív, nem szelektív.
Szelektív intézkedés a gyógyszerek terápiás dózisának a specifikus receptorokra gyakorolt \u200b\u200bhatása. Például a salbutamol hatása a β 2 -adrenoreceptorokra. Ne feledje, hogy a gyógyszerek szelektivitása relatív, az adagok növekedésével eltűnik.
- Helyi, rezorpciós.
helyi a gyógyszer hatása megtörténik, mielőtt felszívódik a vérbe (például kenőcs).
resorptiós A (szisztémás) hatás a gyógyszer vérbe történő felszívódását követően alakul ki. A drogok túlnyomó többségének van ilyen hatása.
Az esetek túlnyomó többségében ahhoz, hogy valamely gyógyszerészeti anyag (ligandum) kifejtse hatását, meg kell felelnie a test meghatározott összetevőinek - célreceptoroknak, molekuláris szerkezeteknek, amelyek fehérje, ritkábban nukleinsavaknak, lipideknek vagy más, a sejtek felületén vagy a felületén elhelyezkedő konfigurációknak, amivel kölcsönhatásba lép, elindítva a biokémiai és fizikai-kémiai folyamatok láncát, amely bizonyos hatást eredményez.
Kétféle membránreceptor létezik - ioncsatornák és receptorok, amelyek a G-proteinhez kapcsolódnak. Például a nátrium-csatorna jellemző az adetilkolinra és hasonló gyógyszerekre. Az acetilkolin kölcsönhatásba lép a csatornafehérjével, konformációs változásokat okozva benne, amelyek hozzájárulnak a csatorna megnyitásához és a nátrium-ionok behatolásához a sejtbe. Ez a folyamat ideges izgalom alatt áll. Egyes gyógyászati \u200b\u200banyagok, amelyek kölcsönhatásba lépnek a nátrium-csatorna fehérjével, megakadályozzák annak megnyitását, gátolva ezzel az ideg gerjesztésének továbbadását.
Az úgynevezett G-protein kapcsolódik a sejtek plazmamembránjának belső részéhez, amely biztosítja a gyógyszer-anyag kölcsönhatási folyamat szinkronizálását a megfelelő intracelluláris célfehérjék egyidejű aktiválásával. Amint az ábrán látható, a gyógyszermolekula kölcsönhatásba lép a membrán külső felületén lévő P-receptorral, ami konformációs változásokat idéz elő a receptor fehérjében. Ennek eredményeként a G-protein megváltoztatja térbeli szerkezetét, a membrán síkjában olyan enzimekre vándorol, amelyek inaktív állapotban vannak a sejtben. A G-protein és az enzimek (T) interakciója határozza meg azok aktiválását (LV / P / T). A noorepinefrin, a dopamin és más ligandumok specifikusan kölcsönhatásba lépnek a G-proteinhez kapcsolódó receptorokkal. Meg kell jegyezni, hogy az acetilkolin nem csak a csatornafehérjével, hanem a G-proteinhez kapcsolódó receptorokkal is kölcsönhatásba léphet.
A ligand és a bioreceptor közötti kölcsönhatás kialakításához szükséges, hogy komplementeritásuk legyen, vagyis közöttük bizonyos affinitásnak vagy affinitásnak kell lennie (a méret megfelelése, térbeli konfiguráció, ellentétes töltések jelenléte stb.). Például egy receptor negatív töltésének meg kell egyeznie egy exogén ligandum pozitív töltésével, és egy anyag nem poláris csoportjai kötődhetnek egy receptor hidrofób helyéhez.
A gyógyszerek fizikai-kémiai tulajdonságai között, amelyek befolyásolják a receptorokkal való kölcsönhatásukat, ki kell emelnünk a molekula méretét, attól függően, hogy az anyag kölcsönhatásba léphet a teljes receptorral vagy annak alkotóelemével. A biológiai membránon keresztüli behatolás kinetikája a gyógyszermolekula méretétől is függ. Jellemzően, amikor egy molekula mérete növekszik, növekszik annak rugalmassága és a van der Waals kötések kialakulásának lehetősége egy makromolekuláris partnerrel. Ezenkívül fontos egy gyógyszermolekula sztereokémiája. A farmakológiai aktivitás a gyógyszer izomer formájától függ. És szem előtt kell tartani: minél meredekebb a receptor molekula konformációja, annál nagyobb a különbség a sztereoizomerek működésében.
A gyógyszer anyag - a receptor kölcsönhatása az intermolekuláris kötéseknek köszönhető. Kezdetben egy anyagot elektrosztatikus erők vonzzák a receptorokhoz, és komplementeritás jelenlétében fizikai és fizikai-kémiai kölcsönhatásokkal (jellemzően azoknak a gyógyszereknek, amelyek változatlan vagy változatlan formában választódnak ki a testből) vagy kémiai kölcsönhatásokkal (a vegyületekre jellemzőek) fizikai és fizikai-kémiai kölcsönhatásokkal kötődnek. a szervezetben kémiai átalakulásokon megy keresztül). A leggyengébb van der Waals-erők részt vesznek egy gyógyszeranyag és a biokémiai reakcióképes rendszerek kölcsönhatásának sajátosságának meghatározásában. A hidrogénkötések részt vesznek az anyag (ligandum) felismerésében és rögzítésében a biostruktúrákhoz. Ionkötések akkor fordulnak elő, amikor a gyógyászati \u200b\u200banyagok kationos vagy anionos csoportot tartalmaznak, és az ellenkező struktúrák a bioreceptorokban vannak. Az anyagok és a receptorok közötti farmakológiai reakció első szakaszában gyakran ionos kötések alakulnak ki. Ilyen esetekben a gyógyszer hatása visszafordítható. Fontos a koordinációs kovalens kötések kialakulása. Részvételükkel az alkilező szerek kölcsönhatásba lépnek a biosubsztrátokkal, valamint a gyógyszerekkel és az antidotumokkal a fémekkel stabil kelátkomplexek kialakulásakor, például az unithiol arzén vagy tetacin-kalcium és az ólom. Az ilyen anyagok hatása visszafordíthatatlan.
Ezen felül hidrofób kölcsönhatás is fennáll. Bár kötéseinek energiája kicsi, számos hosszú alifás lánc kölcsönhatása stabil rendszerek megjelenéséhez vezet. A hidrofób kölcsönhatások szerepet játszanak a biopolimer konformációk stabilizálásában és a biológiai membránok kialakításában.
Az aminosavmaradványok egy fehérjereceptor-molekulában olyan poláris és nem-poláros csoportokat tartalmaznak, amelyek meghatározzák a poláris és nem-poláris kötések kialakulását közöttük és a hatóanyagok között. A poláris csoportok (-OH, -NH, COO-, -N3H, \u003d O) elsősorban ion- és hidrogénkötések kialakulását biztosítják. Nem poláris csoportok (hidrogén, metil, ciklusos csoportok stb.) Hidrofób kötéseket képeznek kis molekulatömegű gyógyászati \u200b\u200banyagokkal.
Így a gyógyszerek kölcsönhatása a specifikus receptorokkal különféle kémiai kötésekkel érhető el, amelyek erőssége egyenlőtlen. Tehát az elektrosztatikus (ionos) interakcióhoz tartozó kolinerg receptorokkal rendelkező kuráraszerű anyagok hozzávetőleges szilárdsága 5 kcal / mol, dipólion - 2-5 kcal / mol, dipól-dipol - 1-3 kcal / mol, hidrogénkötések - 2-5 kcal / mol, van der Waals kötések - 0,5 kcal / mol, hidrofób kötés - 0,7 kcal / CH2 csoport. A kötési szilárdság csökkenése az elektrosztatikus kölcsönhatáshoz használt atomok közötti távolságtól függően r -2, a dipól-ion r -3, a dipól-dipol értéke r -4, a hidrogénkötések r -4, a van der Waals kötések r -7 . Ez a fajta kapcsolat megszakadhat, ami biztosítja a drogok visszafordíthatóságát. Tartósabb a kovalens kötések, amelyek hosszú és gyakran visszafordíthatatlan hatást fejtenek ki az anyagokra, például az alkiláló daganatellenes szerekre. A legtöbb gyógyszer visszafordíthatóan kötődik a receptorokhoz. Ebben az esetben a vegyület természete nagyon bonyolult: az ionos, a dipól-dipól, a van der Waals, a hidrofób és más típusú kötések egyidejűleg részt vehetnek abban, amelyet nagyrészt az anyag és a receptor komplementer jellege, és ennek következtében egymás közötti konvergencia mértéke határoz meg. egyedül.
Egy anyag receptorokhoz való kötődésének erősségét az "affinitás" kifejezés jelöli. Az ugyanazon receptorokon ható anyagok affinitása eltérő lehet. Ebben az esetben a nagyobb affinitással rendelkező anyagok kiszoríthatják az alacsonyabb affinitású anyagokat a receptorokkal szemben lévő vegyülettől. Az "elfoglalt" receptorok (DR), a szabad receptorok és a szabad anyag (D) közötti egyensúlyi állapot meghatározásához a disszociációs állandót (K D) használjuk, amelyet az alábbi képlet határoz meg:
K D \u003d [D] * [R] / [DR]
A KD negatív logaritmusa (pRD) az affinitás mutatója. Az affinitás jellemzésére gyakran alkalmazzák a pD2 mutatót, azaz az EC50 negatív logaritmát (az anyag azon koncentrációja, amelyben a maximális hatás 50% -át okozza).
A kémiai kölcsönhatás-kötés sokféleségét és azok egyenetlen erősségét, vagy affinitását a ligandumok és a bioreceptorok között magyarázza az eltérő reakcióképességű gyököket tartalmazó, sokdimenziós térfogatú alakú gyógyszerek összetett szerkezete, valamint az interakciós folyamatok összetettsége, amelyek gyakran több szakaszban (fázisban) fordulnak elő: komplex kialakulása a gyógyszer anyag egy receptor; intramolekuláris csoportosítás; komplex disszociáció.
Így csak olyan anyagok, amelyek kifejezett affinitással rendelkeznek a bioreceptorral szemben, okozhatnak farmakológiai hatást. A hatás súlyossága a gyógyszer koncentrációjától és a receptorok teljes számától függ.
Ha az anyagoknak elegendő belső aktivitása van, akkor agonistáknak nevezik őket. Belső aktivitással értjük az agonisták azon képességét, hogy biológiai hatást okozzanak a receptorok konformációjának megváltoztatásával, azaz egy ligandum azon képességével, hogy aktiválja a receptort. Ezt a jelenséget az agonista-receptor komplex affinitásának tekintjük az átalakítóhoz; a külső jelek belső jellé történő átalakítását transzdukciónak nevezzük. Az intracelluláris jelátvitel olyan folyamatok alapját képezi, mint az izomrostok összehúzódása, a sejtosztódás, a proliferáció, a differenciálódás stb. Megállapítottuk, hogy a sejtben sok anyag található (hormonok, bioaktív peptidek, nukleotidok, szteroidok, kis molekulatömegű bioregulatorok stb.) specifikus receptorok. Ezen anyagok ezen specifikus receptorokkal való kölcsönhatásának eredményeként másodlagos hírvivők (közvetítők) alakulnak ki, amelyek a biokémiai reakciók kaszkádját idézik elő.
Van egy " részleges agonisták"- gyógyászati \u200b\u200banyagok, amelyek receptorokhoz kötődve nem adják meg a maximális hatást. Ez az érthetetlen jelenség állítólag annak következménye, hogy a gyógyszer-receptor komplex affinitása hiányos (kisebb) függést mutat a tranduktorhoz. Például egy parciális opiát receptor agonista nalorfin hasonlóan működik, mint ezen receptorok teljes agonistája, a morfin, bár ennél gyengébb, mint az utóbbi. Ugyanakkor együttes alkalmazás esetén a nalorfin gyengíti vagy kiküszöböli a morfin hatásait; különösképpen kiküszöbölhető a morfin légzésgátló hatása. Az izoprenalin valódi agonista, a prenalterol pedig a β-adrenerg receptorok részleges agonistája. A receptor elmélet szerint egy igazi agonista maximális választ képes indukálni, még akkor is, ha csak a receptorok egy részével lép kölcsönhatásba.
A specifikus receptorok azonos vagy eltérő kötőhelyekkel rendelkezhetnek az agonisták és antagonisták számára. Különböző kötési helyek lehetnek a különböző agonisták számára. Abban az esetben, ha az agonistán és az antagonistán ugyanazok a kötőhelyek vannak, és az antagonista blokkoló hatása a receptorra teljes mértékben kiküszöbölhető az agonista koncentrációjának növelésével (az agonista maximális hatását elérik), az antagonista és az agonista közötti kapcsolatot verseny antagonizmusnak nevezik. Ha az agonista és az antagonista kötőhelyei eltérőek, akkor a közöttük fennálló kapcsolatot nem-kompetitív antagonizmusnak kell meghatározni. Az antagonisták jellemzésére gyakran alkalmazzák a pA2-t (az antagonista moláris koncentrációjának negatív logaritmusa, amelynél annak koncentrációját meg kell duplázni az agonista standard hatásának eléréséhez).
Egy teljes organizmus körülményei között az agonisták és antagonisták változásokat okoznak bizonyos élettani funkciókban. Ebben az esetben az antagonisták hatását az a tény határozza meg, hogy gátolják a specifikus természetes ligandumoknak a specifikus receptorokra gyakorolt \u200b\u200bhatását (például az atropin M-kolinerg antagonista gátolja azok acetilkolin-agonistájának hatását). Azoknak a változásoknak, amelyek közvetlenül kapcsolódnak az anyagok és a specifikus receptorok közötti kölcsönhatáshoz, az "elsődleges farmakológiai reakció" kifejezéssel jelöljük, amely egy olyan reakciósorozat kezdete lehet, amely bizonyos fiziológiai funkciók stimulálásához vagy gátlásához vezet. "
A szervek vagy rendszerek működésében bekövetkező változásokat (például a szív összehúzódások erősségének és gyakoriságának, a belső szervek simaizom-tónusának, a mirigyek szekréciójának, vérnyomásának stb. Változásait) egy gyógyszer anyag okozza ennek az anyagnak a farmakológiai hatásai. Tehát a szívglikozidok esetében az elsődleges farmakológiai reakció a szívizomszálak Na +, K-ATPáz transzportjának aktivitásának gátlása, amelyet a szívglikozidok lehetséges specifikus receptorának tekintik. Ebben a tekintetben megszakad a K + belépése az izomrostokba és a kilépés az Na + rostokból, és növekszik a citoplazma Ca2 + tartalma, ami elősegíti az aktin és a miozin kölcsönhatását. Ezeknek a változásoknak a következménye a pulzusszám növekedése, amely a szív glikozidjainak fő farmakológiai hatása.
A specifikus receptorok agonistákkal való hosszan tartó expozíciója gyakran kíséri csökkenteni azok érzékenységét. Ez utóbbi kapcsolódhat a receptorok megváltozásához, számuk (sűrűség) csökkenéséhez vagy a receptorok gerjesztését követő folyamatok megszakításához. Ebben az esetben az agonisták farmakológiai hatása kevésbé lesz kifejezett.
Így a legtöbb gyógyszer farmakológiai hatásai a megfelelő specifikus receptorokra gyakorolt \u200b\u200bhatásukhoz kapcsolódnak.
A bioreceptorral szemben nagy affinitású és alacsony belső aktivitású anyagokat antagonistáknak vagy blokkolóknak nevezzük, mivel ezek anélkül, hogy a bioreceptor konformációjában változásokat idéznének elő, akadályozzák az endogén és / vagy exogén agonista ligandumok kölcsönhatását. Vannak úgynevezett "szekunder vagy hülye receptorok, amelyekkel a gyógyászati \u200b\u200banyagok kötődnek, de nincs farmakológiai hatása. Az ilyen "hülye" receptorok leggyakrabban a fehérjékben és a vérplazmában vannak (de a szövetekben is megtalálhatók). A "néma" receptorokkal való kapcsolat a szabad gyógyszeranyag koncentrációjának csökkenéséhez vezet, és ennélfogva a terápiás hatás csökkenéséhez.
Számos modern elmélet magyarázza a ligand-receptor kölcsönhatás mechanizmusát, magának a receptornak az állapotát, az aránytalanság hiányát az elfoglalt receptorok száma és a végső reakció között, a jelátviteli hatékonyság változásait, valamint a tartalék receptorok és parciális agonisták meglétét stb. Alátámasztva. drogcsoportok. Ezeket az interakciókat receptor kölcsönhatásra és kémiai kölcsönhatásra osztjuk.
A gyógyszerek kölcsönhatásának mechanizmusa a bioreceptorral A következő séma szerint ábrázolható: minden ligandum (gyógyászati \u200b\u200banyag vagy fiziológiás szubsztrát) egy adott receptor specifikus helyéhez kötődik. Az aktivált receptorok közvetlenül vagy közvetetten szabályozzák az ionáramot (1) és / vagy más intracelluláris folyamatokat (szekréció vagy izom-összehúzódás), vagy aktiválják a guanin-nukleotid-kötő fehérjék (G-fehérjék) rendszerét, ami viszont fokozza a második enzim-mediátor rendszer aktiválását. Számos különböző második mediátor működik a citoplazmában, aktiválva a különböző célfehérjéket, például a protein-kinázokat. Ez utóbbi specifikus szubsztrátokra hat és közvetíti a farmakológiai hatást.
A bemutatott leírásból látható, hogy a kábítószerek hatását a következő mechanizmusok hajtják végre:
- a szövet fiziológiás funkcióit (például összehúzódó, szekréciós) több receptor, így különféle ligandumok szabályozhatják;
- több köztes lépés lehet egy gyógyszeranyag kölcsönhatása és egy szövet vagy szerv reakciója között, különösen a receptorok második, mediátorral kapcsolatos rendszerének aktiválása között;
- az stimulus-válasz szekvenciáért felelős mechanizmusok hatékonysága, valamint a receptorok sűrűsége szövetekenként változhat.
Bizonyos gyógyszerek terápiás hatása annak közvetlen (nem kapcsolódik a specifikus receptorokhoz) endogén vegyületekkel való kémiai kölcsönhatása vagy más interakciós mechanizmusok (ozmotikus nyomás, adszorpció) következménye. Tehát az ozmotikus diuretikumok - mannit, karbamid - esetében nincs specifikus receptor. Ezek az anyagok növelik a vese tubulusokban az ozmotikus nyomást, ennek eredményeként zavarodik a víz reabszorpciója és növekszik a diurezis. Az adszorbens anyagok, a savképző diuretikumok nem kapcsolódnak a specifikus receptorokhoz.
Antacidok (például alumínium vagy magnézium-hidroxidok) sósavval reagálva gyenge sav tulajdonságokkal rendelkező termékeket képeznek. Bizonyos fémekhez kötő kelátképző szerek inaktív kémiai komplexeket képeznek.
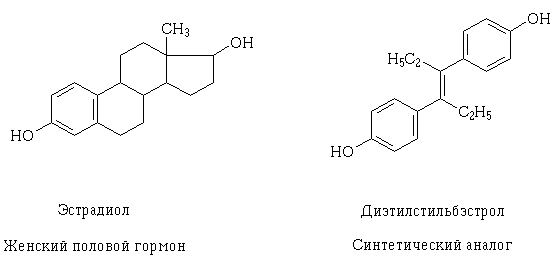
Mivel a receptorok szerkezetével és a gyógyszerek lehetséges farmakodinámiás kölcsönhatásainak sejtszintű ismerete elmélyül, célszerű ezeket létrehozni, valamint elmagyarázni, hogy miért lehet a gyógyszereknek ilyen hatása, amelyek szerkezete első pillantásra különbözik egymástól. Ilyen jelenség például az ösztradiol és a dietil-sztilbestrol transzizomerje, amely a női nemi szintetikus analógja. Szerkezeti molekuláik különböznek, de tulajdonságaikban és méretükben azonos hidroxilcsoportokat tartalmaznak, hasonlóan elhelyezkednek és az űrben orientálódnak, amelynek következtében ezen anyagok molekulái kölcsönhatásba léphetnek ugyanazzal a receptorral, és hasonló farmakológiai hatással rendelkeznek.
Azokat a módszereket, amelyekkel a gyógyhatású anyagok bizonyos farmakológiai hatásokat okoznak, a "hatásmechanizmus" kifejezés jelöli. Ezt a fogalmat magyarázzák a gyógyszerek hatásainak molekuláris, szervi és szisztémás szintjén. Például az antikolineszteráz szerek molekuláris szintű hatásmechanizmusa az acetilkolinészteráz blokádjává redukálódik anionos és észteráz centrumokkal való kölcsönhatás révén. Ugyanakkor az antikolineszteráz drogok vérnyomáscsökkentő mechanizmusának magyarázatát megmutatják, hogy ez a hatás a bradycardia és az értágítás, vagyis megvizsgálják ennek a hatásnak a mechanizmusát a szervek szintjén.
Folyamatban vannak a kábítószerek hatásmechanizmusainak tanulmányozása, és a droganyag hatásmechanizmusára vonatkozó ötletek nemcsak részletesebbé válnak, hanem jelentősen meg is változhatnak az új adatok megszerzésekor.