A túlnyomó többség gyógyszerek van terápiás hatás az evolúció során a testben termelődő sejtek fiziológiai rendszereinek aktivitásának megváltoztatásával. Egy gyógyhatású anyag hatására a szervezetben általában nem merül fel új típusú sejttevékenység, csak a különböző természetes folyamatok üteme változik. A fiziológiai folyamatok gátlása vagy gerjesztése a test szöveteinek megfelelő funkcióinak csökkenéséhez vagy fokozódásához vezet.
A gyógyszerek hathatnak specifikus receptorokra, enzimekre, sejtmembránokra vagy közvetlenül kölcsönhatásba léphetnek a sejtekkel. A cselekvési mechanizmusok részletei gyógyászati \u200b\u200banyagok általános vagy kísérleti farmakológia során tanulmányozták. Az alábbiakban csak néhány példát mutatunk be a kábítószerek fő működési mechanizmusairól.
Hatás specifikus receptorokra. A receptorok makromolekuláris szerkezetek, amelyek szelektíven érzékenyek bizonyos kémiai vegyületekre. A vegyi anyagok és a receptor kölcsönhatása biokémiai és élettani változásokhoz vezet a testben, amelyek egy adott klinikai hatásban fejeződnek ki.
Azon gyógyszereket, amelyek közvetlenül gerjesztik vagy növelik a receptorok funkcionális aktivitását, agonistáknak nevezzük, és olyan anyagokat, amelyek gátolják a specifikus agonisták hatását, antagonistáknak nevezzük. Az antagonizmus versenyképes és nem versenyképes lehet. Az első esetben a hatóanyag egy természetes szabályozóval (mediátorral) verseng a specifikus receptorok kötőhelyein. A kompetitív antagonista által kiváltott receptor blokád nagy adagokban agonista vagy természetes mediátorral kiküszöbölhető.
A receptorok sokféleségét megosztják a természetes mediátorokkal és antagonistáikkal szembeni érzékenység. Például az acetilkolin-érzékeny receptorokat kolinerg, az adrenalin-érzékeny receptorokat pedig adrenerg receptoroknak nevezzük. A muszkarinnal és a nikotinnal szembeni érzékenység szempontjából a kolinerg receptorokat muszkarin-érzékeny (m-kolinerg receptorok) és nikotin-érzékeny (n-kolinerg receptorok) csoportokra osztják. A H-kolinerg receptorok heterogének. Megállapítást nyert, hogy különbségük a különféle anyagok. Vannak n-kolinerg receptorok az autonóm idegrendszer ganglionjaiban, és a csíkos izom n-kolinerg receptorok. Az adrenerg receptorok különféle altípusai ismertek, amelyeket α1, α 2, β1, β2 görög betűk jelölnek.
A H1 és H2 hisztamin, dopamin, szerotonin, opioid és egyéb receptorok szintén elkülönülnek.
Hatás az enzimaktivitásra. Egyes gyógyszerek növelik vagy gátolják a specifikus enzimek aktivitását. Például a fizosztigmin és a neostigmin csökkentik a kolinészteráz aktivitását, amely elpusztítja az acetilkolint, és olyan hatásokat eredményez, amelyek jellemzőek a parasimpatikus idegrendszer gerjesztésére. A monoamin-oxidáz inhibitorok (iprazid, nialamid), amelyek megakadályozzák az adrenalin elpusztulását, növelik a szimpatikus idegrendszer aktivitását. A fenobarbitál és a zixorin, növelve a máj glükuronil-transzferáz aktivitását, csökkenti a vér bilirubin szintjét.
Fizikai-kémiai hatás a sejtmembránokra. Az ideg- és izomrendszer sejtjeinek aktivitása az ionáramlástól függ, amely meghatározza a transzmembrán elektromos potenciált. Egyes gyógyszerek megváltoztatják az ionszállítást.
Tehát antiaritmiás, görcsoldók, általános érzéstelenítésre szolgáló gyógyszerek.
Közvetlen kémiai kölcsönhatás. A gyógyszerek közvetlenül kölcsönhatásba léphetnek a sejten belüli kis molekulákkal vagy ionokkal. Például az etilén-diamin-tetraecetsav (EDTA) erősen köti az ólom-ionokat. A közvetlen kémiai kölcsönhatás elve sok ellenszer használatának alapját képezi a kémiai mérgezésnél. Egy másik példa a sósav antacidokkal történő semlegesítése.
farmakodinámia
Vizsgálja a gyógyszerek hatásmechanizmusát, valamint biokémiai és fiziológiai hatásaikat. Feladatai között szerepel a gyógyszer és a célsejt közötti kémiai és fizikai kölcsönhatások leírása, valamint annak farmakológiai hatásainak teljes spektruma és súlyossága. A farmakodinamikai minták ismerete lehetővé teszi a megfelelő gyógyszer kiválasztását. A farmakodinámiás vizsgálatok mélyebben megismerik a biokémiai és élettani folyamatok szabályozását a testben (Katzung B.G., 1998; Lawrence D.R. et al., 2002).
A legtöbb gyógyszer hatását a test makromolekuláinak való kötődésük közvetíti. Ezen makromolekulák funkcionális állapotának megváltozása viszont biokémiai és fiziológiai reakciók láncolatát indítja el, amelyek farmakológiai hatássá alakulnak. Makromolekulákat, amelyekkel a vegyi anyagok kölcsönhatásba lépnek, receptoroknak nevezzük. Így bármilyen funkcionálisan aktív makromolekula szolgálhat a gyógyszerek receptoraként. Ennek a megállapításnak számos fontos következménye van. Először, gyógyszerek segítségével megváltoztathatja a test bármely fiziológiai folyamatának sebességét. Másodszor, a gyógyszerek csak megváltoztatják a sejt természetes fiziológiai funkcióit anélkül, hogy új tulajdonságokat adnának neki.
receptorok
A legtöbb receptor fehérjék. Ezek a hormonok, növekedési faktorok, mediátorok, proteinek, amelyek részt vesznek a legfontosabb metabolikus és szabályozási reakciókban (dihidrofolát-reduktáz, acetil-kolinészteráz), transzportfehérjék (Na +, K + -ATPáz), szerkezeti fehérjék (tubulin). A különböző kémiai természetű sejtkomponensek, például a nukleinsavak, amelyekkel a daganatellenes szerek kölcsönhatásba lépnek, receptorokként is működhetnek.
Az endogén szabályozó tényezők - hormonok, mediátorok stb. - receptorai farmakológiai jelentőséggel bírnak. Ezek a receptorok számos gyógyszer célpontjaként szolgálnak, általában szelektíven hatnak, mivel a receptorok endogén ligandumok számára nagyfokú specifitást mutatnak. Azokat a gyógyszereket, amelyeket a receptorhoz való kötődés után reprodukálják az endogén ligandum fiziológiai hatását, aganistáknak vagy stimulánsoknak nevezzük. Antagonistáknak vagy blokkolóknak nevezzük azokat a gyógyszereket, amelyek nem idézik elő ezt a hatást, de gátolják az endogén ligandumok kötődését. Azokat az anyagokat, amelyek hatása kevésbé nyilvánvaló, mint az agonisták, részleges agonistáknak nevezzük. Azokat a készítményeket, amelyek stabilizálják a receptort inaktivált konformációban, fordított agonistákként osztályozzák.
Szerkezeti és funkcionális függőség
A gyógyszer kémiai szerkezete meglehetősen mereven határozza meg affinitását a receptorokhoz és a belső aktivitást. A kémiai szerkezet kis változása jelentősen befolyásolja a farmakológiai tulajdonságokat.
Az új gyógyszerek szintézise nagyrészt ezen alapszik. Mivel a kémiai módosítás nem feltétlenül érinti minden farmakológiai tulajdonságot azonos módon, javítható a gyógyszer hatékonysága és biztonsága, növelhető a szelektivitás és javíthatók a farmakokinetikai tulajdonságok. Például sok, a klinikában alkalmazott hormon- és mediátor antagonistát az endogén anyagok kémiai módosításával szintetizálnak.
Kábítószer-alkalmazási pontok
Mivel a gyógyszerek hatását a receptorok közvetítik, a gyógyszer alkalmazásának pontját nem csak eloszlásának jellemzői, hanem a receptorok elhelyezkedése is meghatározza, és a farmakológiai hatások ezen receptorok funkcionális jelentőségétől függenek. Azoknak a gyógyszereknek a farmakológiai hatása, amelyek receptjei sok szervben és szövetekben különböznek. Ha ezek a receptorok a sejtek szempontjából létfontosságú funkciót látnak el, akkor nem csak a gyógyszer terápiás célokra történő felhasználása nehéz, hanem biztonságos is. Ennek ellenére az ilyen gyógyszerek nagy klinikai jelentőséggel bírhatnak. Tehát a szívelégtelenségben széles körben alkalmazott szívglikozidok megváltoztatják az ionok transzportját a sejtmembránon keresztül, amelytől a sejt életképessége függ. Szűk terápiás tartományúak és nagyon mérgezőek. Egy másik példa a daganatellenes szerek. Ha a receptorok, amelyekkel a gyógyszer kölcsönhatásba lép, csak néhány típusú differenciált sejtben vannak jelen, akkor a hatása szelektívebb. Ezeknek a gyógyszereknek kevesebb mellékhatása lehet, ám ezek a gyógyszerek még akkor is mérgezőek lehetnek, ha receptoruk életképes funkciót tölt be. Egyes biológiai méregek (botulinum toxin stb.) Hasonló módon viselkednek. Ezen túlmenően, még ha a közvetlen farmakológiai hatás szelektív is, annak következményei változatosabbak lehetnek.
Endogén szabályozó faktor receptorok
A receptor kifejezés a sejt bármely olyan makromolekuláris komponensére utal, amelyhez a gyógyszer kötődik. Az egyik legfontosabb gyógyszerreceptor a sejtes fehérjék, amelyek endogén szabályozó tényezők - hormonok, növekedési faktorok, mediátorok - receptoraiként szolgálnak. Az endogén ligandumhoz kötődve a receptorok továbbítják a jelet a célsejtbe.
A receptorból a jel közvetlenül a sejtes célpontokon (effektorfehérjék) érkezik, vagy közbenső jelátviteli molekulákon keresztül - protein-átalakítók. A receptor, a protein-átalakítók és az effektorfehérjék képezik a receptor-effektor rendszert. A jelátviteli láncban legközelebbi effektorfehérje gyakran nem terminális effektor (közvetlenül befolyásolja a sejtfunkciókat), hanem egy enzim vagy transzportfehérje, amely részt vesz egy második mediátor - egy ion vagy egy kis molekula - kialakításában, transzportjában vagy inaktiválásában. A második mediátor viszont információkat továbbít számos intracelluláris célponthoz, biztosítva ezzel egyidejű válaszukat az egyik receptor jeleire.
A receptorok, a konvertáló fehérjék és az effektorfehérjék nemcsak az információkat továbbítják. Egyrészt a különféle ligandumokból származó jeleket koordinálják, másrészt ezeket a jeleket a sejtben zajló anyagcsere-folyamatokkal koordinálják.
Katalizátorként működve a receptorok javítják a biológiai szignált. Ennek a fontos tulajdonságnak köszönhetően kiváló célpontokként szolgálnak a gyógyszerek számára. A szignálerősítők azonban nemcsak az enzimatikus aktivitású receptorok, hanem az összes ismert receptor. Valójában, amikor egy ligandum molekula egy ioncsatornával konjugált receptorhoz kötődik, sok ion áthalad az utóbbion. Ugyanez vonatkozik a szteroid hormonreceptorokra: egy hormon molekula kiváltja az mRNS sok példányának transzkripcióját, amelynek alapján számos protein molekulát szintetizálnak.
A biológiailag aktív anyagok receptorait a struktúrától és a hatásmechanizmustól függően több osztályra osztják. Ezen osztályok száma kicsi.
Enzimatikus receptorok
Az enzimatikus aktivitású receptorok legnagyobb csoportja a membránreceptor, amelynek saját protein kináz aktivitása van. Különböző effektorfehérjéket foszforilálnak, amelyek a sejtmembrán belsejében helyezkednek el. Ennek eredményeként ezen fehérjék funkciója vagy más fehérjékkel való kölcsönhatásuk megváltozik.
Van egy másik osztály a protein-kináz aktivitással rendelkező receptorokról - ezek a proteinkinázokkal konjugált receptorok. Hiányzik az intracelluláris katalitikus domén, de amikor egy agonistával kölcsönhatásba lépnek, az membrán belső felületén intracelluláris protein kinázokat kötnek vagy aktiválnak. Ezek a neurotróf faktorok receptorai, valamint a T és B limfociták antigént felismerő receptorai, amelyek több alegységből állnak. Ez utóbbi kölcsönhatásba lép a foszfotirozin-foszfátokkal. Más, az intracelluláris effektor domén nélküli receptorok működését közvetíthetik más effektorfehérjék is.
Más, saját enzimatikus aktivitással rendelkező receptorok hasonló szerkezetűek. Ide tartoznak például a saját foszfotirozin-foszfatáz-aktivitással rendelkező receptorok: extracelluláris doménjük aminosav-sorrendben hasonló a adhéziós molekulákhoz. Sok saját foszfotirozin-foszfatáz-aktivitással rendelkező receptor esetében az endogén ligandumok nem ismertek. A különféle sejttípusokon végzett genetikai és biokémiai vizsgálatok szerint ezeknek a receptoroknak az enzimatikus aktivitása fontos szerepet játszik. A pitvari natriuretic hormon receptorok, más NUP-k, valamint a guanilin receptorok intracelluláris doménje saját guanilát-cikláz aktivitással rendelkezik, és szintetizálja a cGMP-t, amely második mediátorként működik. Talán vannak más receptorok, amelyek saját enzimatikus aktivitással rendelkeznek.
Ioncsatorna kapcsolt receptorok
Egyes mediátorok receptorai közvetlenül kapcsolódnak az ioncsatornákhoz, és kölcsönhatásba lépnek egy ligandummal, hogy szelektíven áthaladjanak bizonyos ionokat a sejtmembránon (kemoszenzitív csatornák, ionotropikus receptorcsatornák, ionotropikus receptorok).
G-proteinhez kapcsolt receptorok
Ez egy olyan meglehetősen nagy osztályú receptor, amely G-fehérjék révén kölcsönhatásba lép az effektorokkal (olyan proteinek, amelyek guanin-difoszfátot (GDF) helyettesítenek guanin-trifoszfáttal (GTP). Ezek ide tartoznak számos biogén amin, a lipid jelző molekulák (különösen az eikoszanoidok) és a különféle peptid receptorai. Az enzimek (adenilát-cikláz, foszfolipáz C), valamint a kálium- és kalciummembrán-csatornák effektorokként funkcionálnak. A G-fehérjékhez kapcsolt receptorok nagy száma és fontos fiziológiai szerepe kiválóvá teszi őket. a gyógyszereim célkitűzései: Az orvosok által felírt összes gyógyszer (az antibiotikumok kivételével) körülbelül a fele hat a fenti receptorokra.
Egy sejt akár 20 receptort hordozhat a felületén, amelyek mindegyike szelektíven kölcsönhatásba lép egy vagy több G-fehérjével (különbözik az α-alegységek különböző típusaitól). Az α-alegység képes kölcsönhatásba lépni egy vagy több effektorfehérjével, ami lehetővé teszi a különböző ligandumok receptorainak jeleinek egy G-fehérje segítségével történő koordinálását. Másrészt, egyetlen receptor képes az intracelluláris szignál átvitel számos mechanizmusát kiváltani, aktiválva többféle G-fehérjét, és ugyanazon a-alegységen keresztül hatással lehet a különböző effektorfehérjékre. A jelek divergenciájának és konvergenciájának ilyen összetett rendszere biztosítja a sejtfunkciók rugalmas szabályozását (Ross, 1992).
Intracelluláris receptorok
A szteroid- és pajzsmirigyhormonok, a kalcitriol és a retinoidok oldódó intracelluláris DNS-kötő fehérjék, amelyek bizonyos gének transzkripcióját szabályozzák (Mangelsdorf et al., 1994). Ezek a receptorok a ligand-érzékeny transzkripciós szabályozók szupercsaládjába tartoznak. A transzkripciós faktorok működését a foszforiláció, a sejtes fehérjékkel, a metabolitokkal és a sejt egyéb szabályozó komponenseivel való kölcsönhatás szabályozza.
Második közvetítő rendszerek
másodlagos közvetítő rendszerek is részt vesznek a külső jelek integrálásában. Bár sokkal több ismert receptor és fehérjejelző molekula van, mint a második mediátorok, az utóbbiak számos sejtjein vesznek részt a sejtjelek átvitelében. A leginkább tanulmányozott második közvetítők közé tartozik a cAMP, cGMP, Ca 2+, IF 3 (inozitol-trifoszfát), DAG (diacil-glicerin), NO. A heterogén vegyületek e csoportja folyamatosan növekszik. A második mediátorok közvetlenül (egymás anyagcseréjének megváltoztatásával) vagy közvetetten (ugyanazon intracelluláris célokra hatva) kölcsönhatásba lépnek. A második mediátorok funkcióját, valamint kialakulásának (vagy felszabadulásának), hasításának és a sejtből történő kiválasztódásának szabályozását célszerűen a cAMP példáján mérlegelni. Ezt a második mediátort az adenilát-citáz hatására szintetizálják, számos G-fehérjékkel konjugált receptor aktiválásakor. A G s fehérje aktiválja az adenilát ciklázt, a G i protein gátolja.
Legalább 10 szövet-specifikus adenilát-ciklotáz-izoform van, amelyek különböznek egymástól az aktivitás szabályozásának mechanizmusaiban.
Általános szabály, hogy a cAMP aktiválja a protein-kinázokat A (cAMP-függő protein-kinázok), a rokon fehérjék kis csoportját. Ezek a protein-kinázok nemcsak a végső intracelluláris célokat (enzimek, transzportfehérjék) foszforilálják, hanem más protein-kinázokat és más szabályozó fehérjéket is. Az utóbbiak tartalmazzák például a transzkripciós faktorokat. Ők felelősek a génátírás transzkripciójának cAMP által közvetített szabályozásáért, és késleltetett cellás választ adnak a szignálra. A protein-kinázok aktiválása mellett a cAMP közvetlenül a kationos membrán csatornákon is hat, amelyek fontos szerepet játszanak, különösen az idegsejtek működésében. Tehát a cAMP-ból származó jel biokémiai változások láncát okozza a célsejtben.
Kalcium. Egy másik jól tanulmányozott második mediátor az intracelluláris Ca 2+. A Ca 2+ -ionok különféle módon lépnek be a citoplazmába: a membráncsatornák mentén (G-proteinektől függő, feszültségfüggő, K + vagy Ca-Ca 2+ által szabályozott), valamint az endoplazmatikus retikulum speciális területein elhelyezkedő csatornákon keresztül, amelyek az IF 3 és a csontvázizomban a membrándepolarizáció eredményeként. A kalcium eltávolítása a citoszol plazmából kétféle módon történik: felszívódik az endoplazmatikus retikulumban vagy kiválasztódik a sejtből. A Ca 2+ sokkal több fehérjére továbbítja a jeleket, mint a cAMP - a sejtek metabolizmusában résztvevő enzimek, protein-kinázok, kalcium-kötő fehérjék. Ez utóbbi kölcsönhatásba lép más végső és közbenső effektorokkal.
Fogyasztói szabályozás
A receptorok nemcsak a fiziológiai és biokémiai funkciókat irányítják, hanem a szabályozás tárgyát képezik. Ezt a szabályozást a makromolekuláik szintézisének és bomlásának szintjén hajtják végre, kovalens kötések kialakulásával más molekulákkal, kölcsönhatásba lépve a szabályozó fehérjékkel és a receptor mozgásával. A konvertáló fehérjéket és az effektorfehérjéket szintén szabályozza. A szabályozó jelek származhatnak az intracelluláris transzmissziós útvonalaktól, amelyeket maga a receptor stimulálása (visszacsatolási mechanizmuson keresztül) aktiválhat, valamint más receptoroktól (közvetlenül vagy közvetve).
A gyógyszerreceptorok hosszú távú stimulálása általában csökkenti a reakciót - ugyanabban a koncentrációban a gyógyszer kevésbé kifejezett hatást vált ki. Ez a deszenzitizációnak, refrakternek, addiktívnak nevezett jelenség fontos szerepet játszik a klinikai gyakorlatban: például hosszan tartó használat Az AD-es betegek kezelésére szolgáló β-adrenerg agonisták csökkentik ezekre a gyógyszerekre adott reakció súlyosságát.
A homológiai deszenzitizáció csak az stimulált receptorokra vonatkozik, és specifikus a ligandumra. Heterológ deszenzibilizációval csökken az egyéb ligandumokkal szembeni reakció súlyossága, amelyek receptorai ugyanazon intracelluláris jelátviteli útvonalon hatnak. Az első esetben a negatív visszacsatolást a receptorra gyakorolt \u200b\u200bhatás biztosítja (foszforiláció, proteolízis, csökkent szintézis), a második esetben a receptoron kívül más, az intracelluláris jelátvitelben részt vevő fehérjéket is befolyásolhat.
Éppen ellenkezőleg: ha a receptorokat nem stimulálják hosszú ideig, akkor fokozódik az agonistákkal szembeni érzékenységük (például ha hosszabb időn keresztül kezelik a β-adrenerg blokkolószert, a propronololt, akkor a β-adrenerg receptorok érzékenysége a β-adrenostimulánsokkal szemben növekszik).
Zavarok a károsodott receptor funkciók miatt
A gyógyszeres érzékenység egyéni különbségein kívül vannak olyan betegségek, amelyeket az intracelluláris jelátvitel mechanizmusának bizonyos komponenseinek diszfunkciója okozza a receptorról az effektorba. A magasan specializálódott receptorok funkciójának elvesztésével a betegség fenotípusos megnyilvánulása korlátozott lehet (például a herék feminizációjával, mely genetikai hiányból vagy az androgénreceptorok szerkezeti hibáiból származik). Ha megsértik a sejtjelek átvitelén belüli egyetemesebb mechanizmust, akkor a betegség tünetei sokkal változatosabbak, mint például a myasthenia gravis és az inzulin-rezisztens diabetes mellitus bizonyos formái esetén, amelyeket az N-kolinerg receptorok és az inzulin receptorok autoimmun diszfunkciói okoznak. Bármely olyan komponens hibái, amelyek számos receptor szignálátvitelében részt vesznek, több endokrin rendellenességet eredményeznek. Példa erre a G fehérjehiány heterozigóta formája, amely minden sejtben aktiválja az adenilát-ciklázt (Spiegel és Weinstein, 1995). Ennek a proteinnek a homozigóta formája valószínűleg halált eredményez.
A receptorok szerkezetében vagy lokalizációjában fellépő zavarok gyengült vagy fokozott reakcióként jelentkezhetnek a gyógyszerre, valamint más nemkívánatos hatásokként.
A génreceptorokat kódoló mutációk képesek megváltoztatni a gyógyszer egyszeri felhasználására adott választ és a hosszú távú kezelés hatékonyságát. Például, a β-adrenerg receptorok olyan hibája, amely felelős a hörgők simaizmok ellazításáért és a légúti rezisztencia szabályozásáért, súlyosbítja ezen receptorok β-adrenoszimulánsokkal szembeni érzékenységének csökkenését az AD betegek hosszú távú kezelése során. Mivel a károsodott receptor működéséért felelős mutációkat azonosítják és a megfelelő géneket klónozzák, lehetséges módszerek kidolgozása az ilyen betegségek kezelésére.
Receptor osztályozás
Hagyományosan a gyógyszerreceptorokat azonosították és osztályozták az ezekre a receptorokra ható szelektív agonisták (stimulánsok) és antagonisták (blokkolók) hatásai és relatív aktivitása alapján. Például, az acetilkolin hatásait, amelyek reprodukálódnak, amikor kölcsönhatásba lépnek a muszkarin alkaloid kolinerg receptoraival, és amelyeket az atropin blokkol, muszkarin hatásoknak nevezzük, és azokat a hatásokat, amelyek a nikotin kolinerg receptorokkal való kölcsönhatás során megjelennek, nikotin hatásoknak nevezzük. A muszkarin és a nikotin hatásait közvetítő receptorokat M és kolinerg receptoroknak nevezzük. Noha egy ilyen besorolás általában nem tükrözi a drogok hatásmechanizmusát, kényelmes a hatásaik rendszerezésére. Valójában az az állítás, miszerint egy gyógyszer bizonyos típusú receptorokat stimulál, ugyanakkor meghatározza ennek a gyógyszernek és az ezen hatásokat fokozó vagy gyengítő hatóanyagok spektrumát. Az ilyen állítások érvényessége azonban megváltozhat a receptorok új típusainak és altípusainak azonosításával, a gyógyszerek kiegészítő hatásmechanizmusainak felismerésével vagy a korábban ismeretlen mellékhatásokkal.
Receptor altípusok
A rendkívül szelektív gyógyszerek egyre növekvő számának megjelenésével egyértelművé vált, hogy a korábban ismert receptorfajták sok altípusba vannak osztva. A molekuláris klónozási módszerek jelentős segítségévé váltak az új receptor altípusok tanulmányozásában, és a rekombináns receptorok előállítása megkönnyítette olyan gyógyszerek létrehozását, amelyek szelektíven hatnak ezekre a receptorokra. A receptorok különböző, de rokon altípusai gyakran (bár nem mindig) kölcsönhatásba lépnek különböző agonistákkal és antagonistákkal. Azok a receptorok, amelyekben nem szelektív agonistákat vagy antagonistákat nem azonosítottak, általában nem egyetlen altípusba tartoznak, hanem ugyanazon receptor izoformáihoz, és különálló altípusok különbözhetnek az intracelluláris jelátvitel mechanizmusában is. Az M1 és M3 kolinerg receptorok például a Gq fehérjén keresztül hatnak, amely aktiválja a foszfolipáz C-t, közvetetten Ca2+ felszabadulást okozva az intracelluláris raktárakból, és az M2 és M4 kolinerg receptorokat a G i fehérjén keresztül, amely gátolja az adenilát-ciklázt. Ugyanakkor a receptorok típusokba és altípusokba sorolását gyakran nem a hatásmechanizmus határozza meg, hanem egy véletlenszerű választás, vagy megalapozott ötletek alapján. Tehát az α 1 -, α 2 - és β-adrenerg receptorok különböznek a gyógyszerekre adott válaszban és a jelátvitelben (aktiválják a G i, G q és G s fehérjéket), bár az α és β-adrenerg receptorok különböző típusúak, és α 1 - és α 2 -adrenoreceptorok - azonos típusú különféle altípusokba. Az α 1 -adrenoreceptor izoformái α 1A, α 1B és α 1D biokémiai tulajdonságaikban kevéssé különböznek egymástól; ugyanez jellemző a β-adrenerg receptorok izoformáz altípusaira (β 1, β 2 és β 3).
A receptor altípusok közötti különbségeket nagyon szelektív gyógyszerek létrehozására használják, például olyan gyógyszerek, amelyeknek ugyanazon a szöveten eltérő hatása van, mivel a receptor altípusokhoz kötődnek, amelyek különböznek az intracelluláris jelátvitel mechanizmusában. Ezenkívül a gyógyszerek szelektíven célozhatnak meg bizonyos sejteket vagy szöveteket, amelyek expresszálják az altípus receptorait. Minél nagyobb a szerek szelektivitása (egy adott szövethez vagy egy bizonyos hatáshoz viszonyítva), annál kedvezőbb a gyógyszer előnyei és a nemkívánatos hatások aránya.
Molekuláris genetikai módszerekkel nemcsak a receptorok különböző izoformáit fedezték fel, hanem az új, korábban ismeretlen receptorokat kódoló géneket is. Ezek közül a receptorok közül sok már be van jelölve egy vagy másik ismert osztályba, és működését a megfelelő ligandumok felhasználásával megvizsgálták. Néhány receptoron azonban még nem találtak ligandumokat.
Ugyanazon receptor különböző izoformáinak felfedezése, amelyeket különböző gének kódolnak (különösen, ha az izoformák nem különböznek egymástól az intracelluláris jelátvitel mechanizmusában, és ugyanazon endogén ligandumokkal kölcsönhatásba lépnek), lehetővé teszi a különböző sejtekben lévő receptorok expressziójának független szabályozását, a test igényeinek megfelelően, különböző életkori időszakok.
Nem receptor által közvetített gyógyszerhatás
Nem minden gyógyszer hat a makromolekuláris struktúrákon - receptorokon keresztül. Egyes gyógyszerek kölcsönhatásba lépnek olyan kismértékű molekulákkal vagy ionokkal, amelyek a testben általában vagy egy vagy másik kóros állapotban vannak. Tehát az antacidok semlegesítik a sósavat a gyomorban. A mesna (a vesék által gyorsan kiválasztódó és a szabad gyököket semlegesítő gyógyszer) bizonyos daganatellenes gyógyszerek aktív metabolitjaihoz kötődik, csökkentve a húgyúti mellékhatások súlyosságát. Számos biológiailag inaktív anyag (például mannit) adagolható olyan mennyiségben, hogy elegendő legyen a biológiai folyadékok ozmolaritásának növeléséhez, és ezáltal megváltoztassa az extracelluláris és intracelluláris folyadékok eloszlását. Ezeknek az anyagoknak köszönhetően fokozható a diurezis, növekszik a bcc, eliminálható az agyödéma. Ezen felül hashajtóként használják őket.
Egyes gyógyszerek integrálódhatnak a sejt alkotóelemeibe és megváltoztathatják funkciójukat az ezeket alkotó anyagokat tartalmazó szerkezeti hasonlóságok miatt. Például a purinek és a pirimidinek analógjait beépítik a nukleinsavakba, és antivirális és daganatellenes szerként használják őket.
AP Viktorov "Klinikai farmakológia"
Általános szabály, hogy a gyógyszerek hatásmechanizmusa azon képességén alapul, hogy bonyolult biokémiai n / vagy biofizikai folyamatokat indítsanak (indítsanak), amelyek végül megváltoztatják és / vagy optimalizálják a célsejt funkcionális aktivitását.
A gyógyszerek a következők révén tudják végrehajtani a szervek és / vagy a célsejtek elleni hatását:
Közvetlen kémiai kölcsönhatás;
Fizikai-kémiai kölcsönhatás a sejtmembránon;
Speciális enzimekre gyakorolt \u200b\u200bhatás;
Hatások a szabályozó génekre;
Hatások a specifikus receptorokra.
Közvetlen kémiai kölcsönhatás LS. A gyógyszerek ezen hatásmechanizmusa meglehetősen ritka, és a sejtön kívül is megvalósítható, például a gyomor vagy a bél lumenében. Lényege abban rejlik, hogy a gyógyszerek közvetlen kémiai reakcióba lépnek olyan molekulákkal és / vagy ionokkal, amelyek a testben normál állapotban képződnek, ha patológiás állapot lép fel. A közvetlen kémiai kölcsönhatás példája a gyomor sósavjának semlegesítésének kémiai reakciója savmegkötő gyógyszerek szedésekor (lásd T. 2., 112. oldal).
A gyógyszerek fizikai-kémiai kölcsönhatása a sejtmembránon. A citoplazmatikus membrán egyik fő funkciója az ioncserék megvalósítása a citoplazma és az extracelluláris környezet között. A transzmembrán ioncserére speciális feszültségfüggő transzmembrán ioncsatornákon keresztül is sor kerülhet - nátrium, kálium, kalcium, klór stb. Egyes gyógyszerek, elérve a sejtmembránt, kölcsönhatásba lépnek ezekkel a csatornákkal és megváltoztatják funkcionális aktivitásukat. Tehát például egy IA osztályú gyógyszer, a kinidin antiaritmiás hatása azon a képességén alapszik, hogy képes blokkolni a Na + -ionok átjutását a transzmembrán nátrium-csatornákon keresztül (lásd T. 2, 35. oldal).
A gyógyszerek hatása a speciális enzimekre. Egy viszonylag kis mennyiségű gyógyszer realizálja farmakológiai hatását, amikor megváltoztatja egyes specializált sejtes enzimek aktivitását. Azokat a gyógyszereket, amelyek növelik a sejtes enzimek aktivitását, enzim induktoroknak nevezzük. Ilyen hatást gyakorolnak például altatók és görcsoldó gyógyszer fenobarbitál, amelyek jelentősen növelik a mikroszomális májenzimek aktivitását. A fenobarbitál és ennek közelében az LS biológiai jelentőségét az alábbiakban vesszük figyelembe.
Azokat a gyógyszereket, amelyek gátolják a speciális enzimek aktivitását, enzimgátlóknak nevezzük. Tehát például a monoamin-oxidáz-gátlók (MAO-k) csoportjából származó antidepresszánsok esetében a pirlindol gyógyszer az antidepresszáns hatását úgy realizálja, hogy gátolja a MAO enzim aktivitását a központi idegrendszerben (lásd T. 1, 294. oldal).
Az acetilkolinészteráz enzim aktivitásának gátlására való képesség az antikolineszteráz gyógyszerek, például a fizosztigmin farmakológiai aktivitásának alapja. Ismeretes, hogy fiziológiás körülmények között az acetilkolinészteráz inaktiválja (elpusztítja) az acetilkolint, egy neurotranszmittert, amely gerjesztést közvetít a parasimpatikus idegrendszer szinapszisában. A fizosztigmin, az acetilkolinészteráz aktivitását elnyomva, elősegíti az acetilkolin neurotranszmitterének parasimpátikus rendszerének szinapsáiban történő felhalmozódást, amelynek eredményeként a parasimpatikus idegrendszer tónusa megemelkedik, ami szisztémás szinten a bradycardia kialakulásával (növekvő vérnyomás, alacsonyabb szintű vérnyomás, BP) növekszik. tanuló stb.
A gyógyszerek visszafordíthatóan és visszafordíthatatlanul kölcsönhatásba léphetnek az enzimekkel. Például az enalapril gyógyszer visszafordíthatóan gátolja az angiotenzin-konvertáló enzim aktivitását, ami különösen a vérnyomás csökkenését vonja maga után, míg a szerves foszfor-mérgező anyagok visszafordíthatatlanul gátolják az acetilkolinészteráz aktivitását.
A gyógyszerek hatása a szabályozó génekre. Jelenleg a tudósok olyan gyógyszereket próbálnak kidolgozni, amelyek felismerik farmakológiai hatásaikat azáltal, hogy közvetlenül befolyásolják a szabályozó gének fiziológiai aktivitását. Ez a tendencia különösen ígéretesnek tűnik azután, hogy az emberi genom szerkezetét 2000-ben megfejtették. Úgy gondolják, hogy a szabályozó gének funkciójának szelektív normalizálása gyógyszerek hatására lehetővé teszi számos betegség, beleértve a korábban gyógyíthatatlanok kezelését is, sikerét.
A gyógyszerek hatása a receptorokra. Mielőtt továbblépnénk a gyógyszerek és a receptorok interakciójának sajátosságaira, tisztáznunk kell, hogy mit értünk a „receptor” kifejezés alatt (latin recept - take, take).
A fiziológia folyamán ismert, hogy a "receptor" kifejezés alatt olyan speciális képződményeket értünk, amelyek képesek érzékelni, átalakítani és továbbítani a külső jel energiáját az idegrendszerbe. Az ilyen receptorokat szenzorosnak nevezzük (lat. Sensus - érzés, érzés, érzékelés).
Az érzékszervi receptorok magukban foglalják a hallás, látás, illat, íz, érzés, szervek receptorait. Ezen szervek szenzoros receptorai az úgynevezett exteroreceptorokhoz tartoznak.
Ha az ősi idők óta ismertek azoknak a szenzoros szerveknek a jelensége, amelyek reagálnak a külső irritáció stimulusokra, akkor a testben található szenzoros receptorok jelenlétét a 19. század közepéig megkérdőjelezték. Első alkalommal az I.F.Pion orosz fiziológus javasolta az ilyen receptorok jelenlétét a testben, aki 1866-ban nyúlkísérlet során az aortairritáció következtében csökkentette a vérnyomást. Ez a felfedezés a test belsejében található receptorok kutatására és tanulmányozására vezetett, és ezeket a receptoreket maguknak interoreceptoroknak nevezték.
A 20. század elejére elegendő számú szenzoros interoreceptort fedeztünk fel, és bizonyították azok fontos szerepét a test élettani funkcióinak szabályozásában.
1905-ben J. Langley bebizonyította, hogy amikor egy gyógyszert feltesznek egy sejtmembránra, akkor farmakológiai hatás alakul ki, ha csak annak egy meghatározott részére alkalmazzák. Ezenkívül ez a hely a sejtfelület teljes területének csak kis részét teszi ki. Ez a megfigyelés lehetővé tette J. Langley arra a következtetésére, hogy a gyógyszerekkel kölcsönhatásba lépő speciális receptor helyek léteznek a sejtmembránon.
A drogok hatásának receptor elméletének megalkotásában azonban a prioritás a német élettárs, P. Ehrlich, aki 1906-ban először vezette be a „receptor” kifejezést, és megfogalmazta azt az állítást, miszerint „a gyógyszer nem működik, ha nem rögzítve van a sejtmembránon”. P. Ehrlich elmélete szerint egy gyógyszermolekulának két funkcionálisan aktív csoportja van, amelyek egyike biztosítja a rögzítését a sejtfelszínen a gyógyszerreceptor régiójában, a második funkcionális csoport kölcsönhatásba lép a receptorral, és összetett biokémiai reakciókat indít el, amelyek megváltoztatják annak (sejt) fiziológiai aktivitását. .
Így már a 20. század elején. nyilvánvalóvá vált, hogy az interoreceptoroknak legalább két osztálya van: szenzoros receptorok, amelyek a belső szervek és a test szöveteinek állapotáról információt továbbítanak a központi idegrendszerre; olyan receptorok jelölése, amelyek kölcsönhatásba lépnek a célsejtek funkcionális aktivitását megváltoztató gyógyszerekkel.
Azonnal meg kell jegyezni, hogy a jövőben a tankönyv szövegében a terminológiában, a gyógyszerreceptorokban és a biológiailag aktív anyagokban, azaz a zavarok elkerülése érdekében, azaz címkézett vagy cytoreceptors. "receptor" kifejezéssel, míg az érzékszervi interoreceptorokat funkcionális aktivitásukat jellemző kifejezéssel jelölik, például: "baroreceptors", "fájdalomreceptorok" stb.
P. Ehrlich felfedezése a gyógyszerreceptorok sejtmembránján alapul szolgált a farmakológiai tudomány, különösen a farmakodinamika fejlődésében, amelynek egyik fő feladata a gyógyszerek receptor-hatásmechanizmusainak tanulmányozása.
Jelenleg nagyszámú sejtreceptor felépítését, egyes biológiailag aktív vegyületekkel való kölcsönhatásuk tulajdonságait tárják fel, amelyek egyrészt lehetővé tették az ismert gyógyszerek hatásmechanizmusainak megértését, másrészt pedig az alapját új, nagyon hatékony gyógyszerek létrehozásához.
Természetesen nehéz elképzelni, hogy az evolúció során az emberi testben különböző szintetikus (kémiai úton előállított) gyógyszerek receptorai alakulnak ki, különösen mivel a modern gyógyszerpiacon bemutatott gyógyszerek túlnyomó többségét az elmúlt 50 évben vagy annál kevesebbel szintetizálták. Bebizonyosodott, hogy a sejt receptor berendezése egy nagyon ősi funkcionális-szerkezeti formáció. Tehát az a- és β-adrenoreceptorok (receptorok, amelyeknek a norepinefrin és az adrenalin kölcsönhatása befolyásolja a sejt funkcionális aktivitását) nemcsak az állati sejtekben, hanem a növényi sejtek membránjain is megtalálhatók, például a növényi nittella sejtjeiben, ahol az a- és β- az adrenorecenterek szabályozzák a protoplazma (sejttartalom) mozgását.
Akkor mi a P. Ehrlich által felfedezett drogok receptora, és miért lépnek kölcsönhatásba velük?
Jelenleg nem kétséges, hogy az úgynevezett gyógyszerreceptorok valójában endogén (a szervezetben előállított) biológiailag aktív anyagok receptorai, amelyek részt vesznek a belső szervek és a test szövetek funkcionális aktivitásának szabályozásában. Az ilyen biológiailag aktív vegyületek közé tartoznak az idegvégződésekből az idegjel továbbításakor felszabadult anyagok, valamint a hormonok, vitaminok, aminosavak stb. Minden endogén biológiailag aktív anyaghoz szigorúan specifikus receptor van. Tehát például a testben termelt biológiailag aktív anyag, az adrenalin képes szigorúan specifikus a- és β-adrenoreceptorokat aktiválni, és a glükokortikoszteroidok - a mellékvesekéreg hormonjai - csak a szigorúan rájuk jellemző glükokortikoszteroid receptorokkal lépnek kölcsönhatásba.
Azok a szintetikus gyógyszerek, amelyek a sejtek receptúrájával kölcsönhatásba lépve, kémiai szerkezetükben észlelik hatásukat, többé-kevésbé hasonlítanak az endogén biológiailag aktív vegyületekhez, amelyek kölcsönhatásba lépnek hasonló receptorokkal. Például, a szintetikus vazokonstriktorok (vazokonstrikciót okozó) gyógyszerek, a fenilefrin kémiai szerkezete közel áll a norepinefrin endogén biológiailag aktív anyagához, ezért, mint a norepinefrin, képes stimulálni az a-adrenoreceptorokat.
Időnként kémiai szerkezetük sajátosságai miatt a gyógyszerek kölcsönhatásba léphetnek nem magával a receptorral, hanem a sejtmembrán szomszédos részével. Mivel ebben az esetben a gyógyszer nem lép kölcsönhatásba magával a receptorral, hanem a sejtmembrán szomszédos részével, nem egy izgalmas vagy blokkoló hatásról beszélnek a receptorra, hanem egy allosztatikus (a görög aliosból egy másik, más) hatásról vagy hatásáról. Ennek eredményeként megváltozhat mind a receptor mellett elhelyezkedő membrán szerkezete, mind a receptor egyes komponensei, ami megváltoztathatja a receptor érzékenységét a rá jellemző biológiailag aktív anyaggal szemben. Azokban az esetekben, amikor a receptor érzékenysége növekszik egy biológiailag aktív anyaggal szemben, a receptor szenzibilizációjáról (latin sensus - érzék) vagy szenzibilizációról (latin sensibilis - érzékenység) beszélnek, és azokban az esetekben, amikor a receptor érzékenysége csökken, desenzibilizációról beszélnek. receptor.
Az alloszterikus hatás sajátossága abban rejlik, hogy az ilyen típusú hatásmechanizmussal rendelkező gyógyszerek nem közvetlenül érintik az idegimpulzus átadását, hanem módosítják azt a kívánt irányba. Például a szorongásgátló szerek (szorongásgátló szerek; szinonimája: trankvilizátorok), amelyek kémiai szerkezetükben benzodiazepin származékai, működési mechanizmusa a posztszinaptikus benzodiazepin receptorok alloszterikus gerjesztésének jelenségén alapul. Ez utóbbi gerjesztése elősegíti a gamma-amino-vajsav gátló posztszinaptikus receptorok (GABA-receptorok) aktiválását, amely klinikailag a neurotikus betegségek tüneteinek, például szorongás, szorongás, félelem stb. Megszüntetésével nyilvánul meg.
A receptorokat, amelyek kölcsönhatásba lépnek és amelyekkel egy biológiailag aktív anyag vagy gyógyszer bármilyen módon megváltoztatja a célsejt funkcionális állapotát, specifikusnak nevezik.
A specifikus receptorokon kívül úgynevezett gyógyszer-specifikus receptorokat is izolálnak. A szakorvosi szakirodalomban ezeket a receptorokat a drogok "veszteségének" helyének is nevezik. Ha ilyen gyógyszerekkel érintkeznek, a gyógyszereknek nincs biológiai hatása, de maguk biológiailag inaktívvá válnak. Az ilyen típusú receptor például a plazmafehérjékben, különösen a vízben oldódó fehérjékben - az albuminban található receptorokként szolgálhat. Ennek a jelenségnek a jelentőségét az alábbiakban részletesebben tárgyaljuk (lásd T. 1, 72. oldal).
A receptorok szerkezete meglehetősen bonyolult, de ezek többsége protein makromolekulák vagy glikoproteinek, amelyek tartalmazhatnak ionokat, lipideket, nukleinsavakat stb. Receptor, azaz az azt alkotó fehérje makromolekulát egy specifikus, az egyes receptorokra specifikus, kémiai csoportjainak térbeli elrendezése jellemzi. A receptort alkotó makromolekulát integrálhatjuk (elmeríthetjük) a citoplazmatikus membrán lipid kettős rétegében vagy lokalizálhatjuk a sejt belsejében. A sejtreceptor fő funkciója egy endogén biológiailag aktív anyagon és / vagy gyógyszereken keresztül továbbított kémiai jel „felismerése”, és a sejt megfelelő biokémiai és / vagy biofizikai válaszává történő átalakítása.
Korábban azt hitték, hogy a gyógyszerek vagy az endogén biológiailag aktív anyagok kölcsönhatásba lépnek a „kulcs és zár” típusú receptorokkal, azaz a receptornak olyan szerkezete van, amely lehetővé teszi a gyógyszer számára, hogy megtalálja az „Ön” receptort, csatlakozzon hozzá, és minthogy „bekapcsolja” és „kapcsolja ki”. Az orvostudomány fejlődésével azonban nyilvánvalóvá vált, hogy az ego nem egészen így van. Jelenleg az extracelluláris szignálok intracelluláris, sejtfunkciót szabályozó molekuláris átalakításának molekuláris folyamatait már elég jól megvizsgálták. mechanizmusok, amelyek hatással vannak az endogén biológiailag aktív anyagok vagy gyógyszerek receptorokkal való kölcsönhatására.
Amikor kölcsönhatásba lép egy endogén biológiailag aktív anyag és / vagy egy hasonló aktív L C receptorával, konformáció lép fel - egy fehérje makromolekula formájának térbeli változása, amely kiváltja a különböző intracelluláris folyamatokat, amelyek meghatározzák a célsejt meditátorra és / vagy gyógyszerre adott válaszát. Például, a bronchiális simaizom-adrenerg receptorok aktiválása a β 2 -adrenostimulátor fenoterol hatására az adenilát-cikláz enzim aktivitásának növekedéséhez vezet, amely hozzájárul a ciklikus adenozin-monofoszfát (cAMP) felhalmozódásához a sejtben és ennek eredményeként a sejtek relaxációjához.
Általános biológiai értelemben a celluláris receptorokat szigorúan specializált sejtek „szenzoros szervének” lehet tekinteni, amelyek révén érzékelik például a központi idegrendszerből és / vagy az endokrin rendszerből származó „információt”. A receptorkészülék fontos szerepe ellenére a receptorok a sejtmembrán csak jelentéktelen részét foglalják el. Például egy sejt M-kolinerg receptor berendezése a felületének legfeljebb 1/6 000-ét foglalja el.
A gyógyszerek és a receptor közötti kölcsönhatás jellemzőinek tanulmányozása egyrészt lehetővé teszi, hogy megértsük annak molekuláris mechanizmusának alapjait, másrészt információt nyújt arról, hogy milyen változtatásokat kell végrehajtani a gyógyszerek szerkezetében annak érdekében, hogy javítsák a kölcsönhatás képességét ezzel a receptorral, azaz . lehetővé teszi új, nagyon hatékony gyógyszerek célzott szintézisét.
Fiziológiai körülmények között a különböző celluláris receptorok nem működnek egymástól függetlenül, hanem állandó kölcsönhatásban vannak egymással, ezáltal szabályozva a sejt fajlagos aktivitását. Például, ha a szív β-adrenerg receptorokat endogén norepinefrinnel aktiválják, különösen a szív összehúzódások számának növekedése, és a szívsejtek M-kolinerg receptorjainak endogén acetilkolin általi aktiválása, éppen ellenkezőleg, csökkenti a szív összehúzódások számát.
A pre- és posztszinaptikus receptorok felfedezése nagyban hozzájárult a gyógyszerek receptor-hatásmechanizmusainak megértéséhez. A szinapszis (görög szinapszisból - kapcsolat, kapcsolat) egy speciális kapcsolattartó zóna az idegsejtek vagy a test más ingerlékeny szerkezete között, amely biztosítja a beérkező információk továbbítását és információs jelentőségének megőrzését. A szinapszis szerkezetének és funkcionális szerepének tanulmányozása a 19. század végén kezdődött. ezt követően a S. Ramon n Cajal spanyol histológus (S. Ramon a Cajalnál) egy speciális átviteli rendszer jelenlétét javasolta a központi idegrendszerben. A szinapszis 1897-ben kapta nevét, amikor az angol fiziológus C. Sherrington javasolta, hogy ez a kifejezés az idegsejtek közötti érintkezési területre utaljon.
Jelenleg háromféle szinapszis létezik:
1) „elektromos” szinapszis, amelyben az információ továbbítódik egy preszinaptikus membránról származó elektromos jel átvitelével. Ezt a fajta szinapszist efaps-nek hívják (a görög nyelvből. Efázis - szoros kapcsolat);
2) "kémiai" szinapszis, amelyben az információ speciális biológiailag aktív anyagokon - neurotranszmitterekön keresztül - kerül továbbításra (görög. Neuron - ideg és latin. Közvetítő - közvetítő; szinonimája: mediator);
3) „vegyes” szinapszis, amelyben az információt mind kémiai, mind elektromos úton továbbítják.
A szinapszis működését befolyásoló gyógyszerek túlnyomó többségének farmakológiai hatásai a gótra gyakorolt \u200b\u200bhatásuk vagy a jel átvitelének a kémiai szinapszisban egy másik szakaszában jelentkeznek, azaz a második típusú szinapszisban.
A kémiai szinapszisokat általában az alábbiak szerint osztályozzák az idegimpulzusokat közvetítő neurotranszmitterek:
A szinapszisokat, amelyekben az acetilkolin mediátorként működik, kolinergnek nevezzük;
A szinapszisokat, amelyekben az adrenalin és a norepinefrin mediátorként működnek, adrenergnek nevezzük;
A szinapszisokat, amelyekben az ATP és az adenozin mediátorként működnek, purinergnek nevezik;
Azokat a szinapszákat, amelyekben a gamma-amino-vaj mediátorként működik, GABA-ergikusnak nevezik stb.
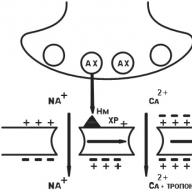
A szinapszis felépítése jelenleg jól ismert. A szinapszis egy idegsejt (axonvég) preszinaptikus folyamatából és egy “jelet” fogadó készülékből áll, amely az effektor („végrehajtó”) sejt membránján helyezkedik el.
Az efferens neuron axonja, amely megközelíti az effektorsejtet, elveszíti a mielinhüvelyt, kitágul és kialakítja az úgynevezett presynapticus vastagodást (1.5. Ábra). Az effektor sejt membránjával szemben levő ideg végét preszinaptikus membránnak nevezzük. Az effektorcellának a preszinaptikus membránnal szemben elhelyezkedő helyét posztszinaptikus membránnak nevezzük (lásd az 1.5. Ábrát). A szinapszis szerkezeti tulajdonságaitól függően az presinaptikus membrán többé-kevésbé redő lehet, így nagyobb vagy kisebb területe van. A kémiai szinapszisban az presinaptikus membrán nem közvetlenül érintkezik a posztszinaptikus membránnal, hanem egy kis távolságra elválasztja tőle, úgynevezett szinaptikus hasadékot (lásd az 1.5. Ábrát).
Presynaptikus sűrűsödés, azaz az axon végrésze nagyobb számú mitokondriumot, az energia szintézisében és felhalmozódásában részt vevő intracelluláris organellákat tartalmaz, ami nagyobb, mint a neuron testénél, ami jelzi az idegsejt ezen szakaszában zajló energiafolyamatok intenzitását. A mitokondriumok mellett az presisznaptikus sűrűsödések nagy számban tartalmaznak kis vezikulákat - vezikulumokat. Egy presisznaptikus megvastagodásban átlagosan körülbelül 20 000 vezikulum található meg. Ez utóbbiak a preszinaptikus vastagodásban egyenetlenül helyezkednek el, általában ezek többsége az presinaptikus membrán közelében helyezkedik el. A neurotranszmittert a neuron testében és axonjában szintetizálják, és felhalmozódnak a vezikulákban. Mindegyik vezikulum több ezer (1 000 - 50 000) neurotranszmitter molekulát tartalmaz. Egy idegimpulzus bekövetkezésekor a vezikulum összeolvad a preszinaptikus membránnal, és a neurotranszmitter kiválasztódik a szinaptikus hasadékba (lásd 1.5. Ábra).
Ábra. 1.5. A "kémiai" szinapszis szerkezetének vázlatos rajza:
a vázlatos kép; b - elektronikus mikrográf; 1- presynapticus idegvégződés; 2 - prsszinaptikus membrán; 3 - posztszinaptikus membrán; 4 - szinaptikus hasadék; B - vezikulum; NM - neurotranszmitter; P - posztszinaptikus receptor: OZ - egy neurotranszmitter "fordított" elfogása; Az SF egy speciális enzim, amely elpusztítja a felesleges neurotranszmittert a szinaptikus hasadékban
A funkcionálisan aktív receptor képződmények a posztszinaptikus membránon helyezkednek el, amelyek képesek kölcsönhatásba lépni a preszinaptikus membránból felszabadult neurotranszmitterrel az idegimpulzus áthaladása során. A posztszinaptikus membránon elhelyezkedő receptorokat szinaptikus vagy posztszinaptikus receptoroknak nevezik a szakorvosi szakirodalomban. A posztszinaptikus receptorok alatt a posztszinaptikus membránba ágyazott, természetű makromolekulákat értjük, amelyek genetikailag előre meghatározott szerkezettel és funkcióval rendelkeznek, és amelyek az aktív központ funkcionális csoportjai (a makromolekula „felismerő” része) miatt visszafordítható kölcsönhatásba lépnek neurotranszmitterekkel és / vagy gyógyszerekkel.
Az idegjel továbbítása a szinapszisban az alábbiak szerint történik: idegi stimulus hatására a vezikulák az presinaptikus membránra mozognak, és az idegtranszmitter az exocitózis által a szinaptikus hasadékba választódik ki (lásd az 1.5. Ábrát). A szinaptikus hasadékba engedő neurotranszmitter eléri a posztszinaptikus membránt, ahol a posztszinaptikus receptorral kölcsönhatásba lépve biokémiai és / vagy biofizikai reakciók láncolatát indítja el, amelynek eredményeként a célsejt élettani reakciója alakul ki. Ugyanakkor a felszabadult neurotranszmitter nem minden mennyiségben jut el a posztszinaptikus receptorokhoz és kölcsönhatásba lép velük. A neurotranszmitter egy részét a presynapticum membrán megragadja, és “visszatér” a tárolóhelyekre. Ezt a jelenséget neurotranszmitter újrafelvétel jelenségnek nevezzük.
A nem kölcsönhatásba lépő neurotranszmitter receptor fennmaradó mennyiségét a szinaptikus hasadékban speciális enzimek pusztítják el. Ezt a jelenséget neurotranszmitterek lebomlásának nevezik. Például az acetilkolinészteráz enzim katalizálja (felgyorsítja) a degradáció (pusztulás) folyamatát az neurotranszmitter acetilkolin szinaptikus hasadékában.
A neurotranszmittertől eltérően, metabolikus termékei neurotranszmitter aktivitással rendelkeznek. A neurotranszmitter és a receptorok közötti interakció teljes folyamata és a felesleg egy speciális enzim általi elpusztítása rendkívül rövid, és nem haladja meg a 2 ms-ot (1 ms \u003d 0,001 s).
Ennek a folyamatnak a ilyen rövid időtartama egyrészt azzal magyarázható, hogy a neurotranszmitter rendkívül gyorsan felszabadul a receptorról, másrészt pedig a szinaptikus kórokozóban a neurotranszmitter enzimatikus inaktiválódásának nagy sebessége magyarázható.
A szinapszis alapvetően funkcionális aktivitása az alábbiak szerint változtatható meg:
A preszinaptikus véget érő neurotranszmitter szintézisének, felhalmozódásának és / vagy katabolizmusának (pusztulásának) felgyorsítása, csökkentése vagy blokkolása. Ennek eredményeként a neurotranszmitter tartalma és ennek következtében fiziológiai aktivitásának intenzitása valamilyen módon megváltozik.
Például a szimpatolitikus rezerpin megakadályozza a katecholaminok felhalmozódását a szinaptikus hólyagokban, teljes kiürülésükig. Ennek eredményeként a szinaptikus hasadékba felszabaduló neurotranszmitter norepinefrin mennyisége hirtelen csökken. Rendszer szinten ez a hatás a vérnyomás csökkenése formájában valósul meg. Egyes gyógyszerek nem közvetlenül befolyásolják a preszinaptikus végződésben levő neurotranszmitterek tartalmát, hanem gátolják azokat elpusztító enzimek aktivitását. Így számos antidepresszáns hat. Például az antidepresszáns pirlindol gátolja (elnyomja) a monoamin-oxidáz enzim aktivitását a preszinaptikus terminációban, és ennek eredményeként növeli a neurotranszmitterek, például norepinefrin, dopamin és szerotonin koncentrációját benne. Klinikailag a pirlindol e hatása a szorongás és félelem csökkenésével, a jobb hangulatban, a megnövekedett fizikai aktivitással stb .;
Megváltoztathatja (megkönnyíti, bonyolítja) a neurotranszmitter képességét az presinaptikus membránon történő áthatolásra, és így minden impulzus mellett növelheti vagy csökkentheti a szinaptikus hasadékba szabaduló neurotranszmitter mennyiségét.
Például a pszichostimuláns amfetamin megkönnyíti a katecholaminok felszabadulását a központi idegrendszer adrenerg szinapszisában, és ezáltal növeli azok tartalmát a szinaptikus hasadékban. Klinikai szempontból a gyógyszernek ez a hatása javul a hangulatban, az erő növekedésének érzése, a megnövekedett teljesítmény. A tetanusz toxin gátolja a gátló neurotranszmitterek (GABA, glicin) felszabadulását a központi idegrendszerben, és ezzel jelentősen csökkenti azok szinaptikus hasadékban lévő tartalmát, amelyet klinikailag a rohamok kialakulása mutat;
Blokkolja vagy stimulálja a neurotranszmitterek újbóli felvételét a preszinaptikus membránon, és ezért növelje vagy csökkentse a neurotranszmitterek koncentrációját a szinaptikus hasadékban.
Például a triciklusos antidepresszáns imipramin blokkolja a norepinefrin neurotranszmitter újbóli felvételét az presinaptikus membránon, és ezáltal meredeken növeli annak koncentrációját a szinaptikus hasadékban. Klinikai szempontból az imipramin ez a hatása a jobb hangulatban, a fokozott mentális és fizikai aktivitásban nyilvánul meg;
Stimulálja vagy blokkolja azon enzimek aktivitását, amelyek elpusztítják a szinaptikus hasadékban a neurotranszmittert.
Például az antikolineszteráz-gyógyszer fizosztigmin csökkenti az acetilkolinészteráz enzim aktivitását, amely elpusztítja a szinaptikus hasadékban az acetilkolint a neurotranszmitterből, és ezáltal hozzájárul annak koncentrációjának növekedéséhez, amely klinikailag megnyilvánulhat, különösen a szem intraokuláris nyomásának és a pupilla összehúzódásának csökkentésével.
Stimulálja vagy blokkolja a posztszinaptikus receptorokat, azaz utánozzák vagy blokkolják a neurotranszmitterek hatását.
Például olyan narkotikus fájdalomcsillapítók, amelyek gerjesztik a postsynapticus opioid receptorokat, és utánozzák a neurotranszmitterek - enkefalinok - hatását. A sztrichin a gátló neurotranszmitter glicin receptorának blokkolásával gátolja annak gátló hatásának megvalósulását, ennek eredményeként a nagy adagokban alkalmazott sztrichin rohamokat okoz.

Ábra. 1.6. A pre- és posztszinaptikus receptorok lokalizációjának vázlatos ábrázolása, amelyet az adrenerg szinapszis mutat be (magyarázat a
NM - neurotranszmitter; M 2 (-) - kolinerg "gátló" preszinaptikus heteroreceptor; β 1 (+) - adrenerg „aktiváló” preszinaptikus autoreceptor; β - adrenerg posztszinaptikus receptor
A posztszinaptikus membránon elhelyezkedő receptorok mellett, azaz posztszinaptikus receptorok, az presszinaptikus membránon elhelyezkedő receptorok, azaz presynatikus receptorok (1.6. ábra). Annak ellenére, hogy mind a pre-, mind a posztszinaptikus receptorokat ugyanaz a neurotranszmitter gerjesztheti, ezeknek a képződményeknek a szinapszisokban funkcionális szerepe eltér. Ha a posztszinaptikus receptorok képezik az idegimpulzus effektor szervbe történő továbbításának végső kapcsolatát, azaz biztosítja az idegimpulzus egyirányú vezetését a központtól a perifériáig, majd az presinaptikus receptorok részt vesznek a
a szinapszis neurotranszmitter aktivitásának szabályozása, azaz bizonyos mértékben befolyásolhatják a benne lévő neurotranszmitter felszabadulási és / vagy szintézis folyamatait. Hangsúlyozni kell, hogy az presinaptikus receptorok közvetlenül nem vesznek részt az idegimpulzusnak az idegtől az effektor szervig történő vezetésében.
A preszinaptikus receptorokat két nagy csoportra osztják: auto- és heteroneuromodulációs receptorok (lásd 1.6. Ábra).
A preszinaptikus autoreceptorok olyan receptorokat foglalnak magukban, amelyeket saját neurotranszmittereik izgatnak erre a szinapszisra.
Például olyan szinapszisokban, amelyek a szomatikus idegek és a csíkos izom érintkezési területén vannak, amikor a neurotranszmitter felesleges mennyiségű acetilkolint tartalmaz a szinaptikus hasadékban, kölcsönhatásban áll a preszinaptikus autoreceptorokkal, gátolja a neurotranszmitter új részének felszabadulását. az presinaptikus autoreceptorok gerjesztése szabályozza az acetilkolin felszabadulását az presinaptikus terminálokból.
Az presinaptikus membránon azonban az autoreceptorok mellett, azaz Megtalálhatók olyan receptorok, amelyek érzékenyek egy adott szinapszisban gerjesztést közvetítő neurotranszmitterre, olyan receptorok, amelyek nem érzékenyek egy adott szinapszisban gerjesztést közvetítő neurotranszmitterre, de más típusú neurotranszmitterrel kölcsönhatásba lépnek.
Például a szinaptikus presinaptikus membránon, amelyben az acetilkolin a neurotranszmitter, a norepinefrin neurotranszmitterre érzékeny presiszinaptikus receptorok helyezkedhetnek el. Ezt a presisznaptikus receptort heteroneuromoduláló receptornak nevezik.
Így a szinapszis egy komplex anatómiai és funkcionális formáció, amely biztosítja az idegimpulzus átvitelét a neuronból a neuronba vagy az idegsejtből az effektor sejtbe.
A szinapszis funkcionális aktivitásának sorrendje (a szinaptikus átadás szakaszai) a következő:
Neurotranszmitter szintézise és felhalmozódása preszinaptikus sűrűsödésekben lokalizált vezikulumokban (egy neurotranszmitter szintézise nemcsak presszinaptikus vastagodásokban, hanem egy idegsejtben és az axonokban is előfordul);
A neurotranszmitter felszabadulása a szinaptikus hasadékba egy idegimpulzus áthaladásakor;
A neurotranszmitter kölcsönhatása a posztszinaptikus receptorokkal, ami magában foglalja a receptorok aktiválását és az effektor sejt funkcionális aktivitásának megváltozását;
A neurotranszmitter inaktiválása (enzimatikus) és / vagy visszavételét az presinaptikus membrán, azaz a szinapszis azon képességének helyreállítása, hogy ismét továbbadja az idegimpulzust az effektor sejtbe.
A szinapszisok a következő alapvető tulajdonságokkal rendelkeznek:
A gerjesztés egyoldalú vezetése (az idegimpulzus csak az presinaptikus membrántól a posztszinaptikusig terjedhet);
Szinaptikus késés, azaz egy bizonyos időt töltünk egy idegi impulzus átvitelére a szinapszisban. (A szinaptikus transzmisszió sebessége átlagosan több mint tízszeresére csökken, mint egy idegimpulzusnak az idegen keresztüli terjedésének sebessége. Kémiai szinapszis esetén általában 0,2 -0,5 ms-ig terjed);
Fáradtság - egy idegimpulzus átvitelének fokozatos csökkenése vagy teljes leállása hosszantartó idegstimulációval. Ennek a jelenségnek az alapja egyrészt a neurotranszmitter-tartalék kimerülése preszinaptikus megvastagodások esetén, másrészt pedig a neurotranszmitterrel szembeni posztszinaptikus receptorokkal szembeni érzékenység csökkentése;
A szinaptikus képződmények magas érzékenysége a gyógyszerekkel és a méreggel szemben.
A szinapszis utolsó tulajdonságán alapul azon gyógyszerek teljes farmakológiája, amelyek befolyásolják a test különféle szerveiben és szöveteiben található szinapszisok funkcionális aktivitását. Hangsúlyozni kell, hogy a farmakológiai hatás tárgya lehet a szinaptikus átvitel bármelyik stádiuma. A szinaptikus átvitelt befolyásoló gyógyszerekként a neurotranszmitterek exogén analógjait, ezek kémiai prekurzorai és más biológiailag aktív anyagokat használnak, amelyek bármilyen módon megváltoztathatják a szinapszis funkcionális aktivitását.
Meg kell jegyezni, hogy sok gyógyszernek nem csak egy, hanem több hatása van a hatásnak a szinapszis szintjén. Tehát például az antidepresszáns pirlindol nem csak gátolja a monoamin-oxidáz enzim aktivitását a szinaptikus hasadékban, hanem blokkolja a norepinefrin újbóli felvételét az presinaptikus membránon.
A receptornak a szinapszishoz való lokalizációja kapcsán preszinaptikus, posztszinaptikus és extrasynaptikus csoportokra lehet osztani. Az utóbbi például a vérlemezkék sejtmembránján elhelyezkedő receptorokat foglalja magában.
A sejt topográfia (elhelyezkedés) szempontjából a receptorokat az alábbiak szerint lehet besorolni a sejtszerkezeten belüli elhelyezkedésük alapján:
membránreceptorok - a citoplazmatikus membránon elhelyezkedő receptorok;
citoszolos receptorok - intracelluláris képződményeken elhelyezkedő receptorok;
nukleáris receptorok - a sejtmag membránján elhelyezkedő receptorok.
Mint korábban megjegyeztük, az endogén biológiai anyagok vagy gyógyszerek receptorával való kölcsönhatás eredményeként a célsejtek funkcionális aktivitása megváltozik. Ez a folyamat különféle módon valósítható meg, szigorúan meghatározva a különféle receptorok számára. Ennek megfelelően jelenleg négyféle receptort különböztetünk meg, amelyek mindegyikének megvan a sajátja, amely alapvetően különbözik a többitől, és amelynek révén a receptorból származó jel biokémiai és / vagy biofizikai reakciók kaszkádját indítja el, amely megváltoztatja a célsejtek funkcionális állapotát.
Az első három típusú receptor a sejt (citoplazmatikus) membránon helyezkedik el, a negyedik típusú receptorok citoszolos és nukleáris receptorokat tartalmaznak.
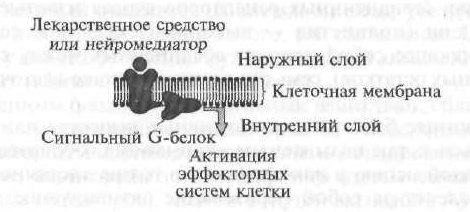
Az I. típusú receptorok olyan celluláris (membrán) receptorokat foglalnak magukban, amelyek hatásukat az úgynevezett jelző G-proteinek révén realizálják (1.7. Ábra).
Az első szakaszban egy biológiailag aktív anyag vagy gyógyszer, amely "felmegy" a sejtmembránhoz, "felismeri" a receptort és kölcsönhatásba lép vele, majd a receptor aktiválja a membrán belső felületén található speciális G jelű fehérjét. Ezenkívül az aktivált G-protein megváltoztatja a belső effektor elem funkcionális aktivitását, amely rendszerint enzimek. Ezután az effektor elem, amely enzim, aktiválja a szekunder hírvivőt vagy egy szekunder hírvivőt, amely olyan biokémiai reakciók sorozatát indítja el, amelyek megváltoztatják a célsejtek funkcionális aktivitását.
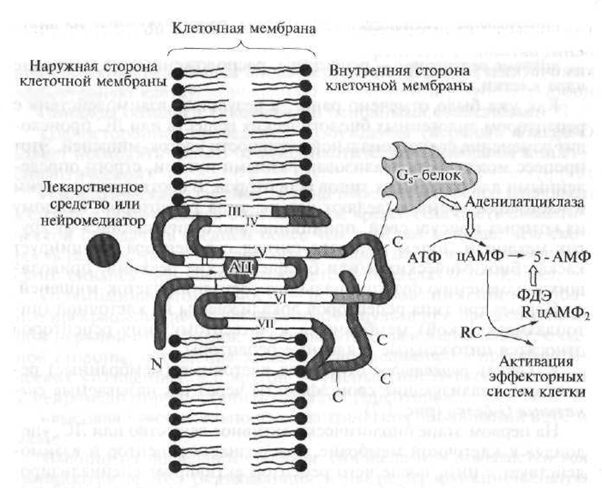
I. típusú sejtreceptorok, azaz A szignál G-fehérjékkel konjugált receptorok szerkezetileg hasonlóak egymással, és térbeli felépítésükben szerpentin (francia szerpantinból - kígyó, labda) szerkezetűek (1.8. ábra).
Ábra. 1.7. Az 1. típusú receptor szerkezete (magyarázat a szövegben)
Ábra. 1.8. A "szerpentin" szerkezetének vázlatos ábrázolása
receptor:
N a receptor polipeptid része, amely a sejtmembrán felett helyezkedik el; C a receptor polipeptid része, amely a sejtmembrán alatt helyezkedik el; AC - a receptor aktív központja, amellyel a gyógyszer kölcsönhatásba lép; ATP - adenozin-trifoszfát - másodlagos hírvivő; cAMP - ciklikus adenozin-monofoszfát; 5-AMP - adenozin-5 "monofoszfát; PDE - foszfodiészteráz; R. RC - cAMP-függő enzim (protein kináz) szabályozó és katalitikus (a reakciót gyorsító) alegységekkel; 1-VII - a szerpentin receptor polipeptid láncai
A szerpentinreceptorok körvonalazott polipeptidláncokat tartalmaznak (a polipeptid nagy molekulatömegű vegyület, amely összekapcsolt aminosavmaradék-lánc), amelyek hétszer áthatolnak a sejtmembránon.
Az endogén biológiailag aktív anyagok vagy gyógyszerek kötődhetnek a polipeptidlánc által alkotott és a sejtmembrán vastagságában lévő úgynevezett „zsebhez”, amely aktiváló jel kialakulását vonja maga után, amelyet a sejt citoplazmájában elhelyezkedő receptorlánc részeire továbbítanak. A szignál G fehérjék kölcsönhatásba lépnek a ci-
a padló és a peptidlánc stol (intracelluláris) szakaszai; aktiválni és elindítani egy olyan biokémiai reakciók kaszkádját, amelyek megváltoztatják funkcionális aktivitását a célsejt körül, azaz elsődleges farmakológiai választ indíthat.
Jelenleg számos G típusú szignálfehérje ismert.
G jel, -proteinek. Ezek a jelző fehérjék rendszerint effektor elemet - az adenilát-cikláz enzimet aktiválnak -, amely serkenti a szekunder hírvivő - ciklikus adenozin-monofoszfát (cAMP) szintézisét (ATP-ből) a sejtben. A cAMP mint másodlagos hírvivő biológiai szerepe nagyon fontos. Például, ha a tartalma növekszik a szívsejtekben, akkor a szív összehúzódásainak gyakorisága és erőssége növekszik. Ezenkívül a cAMP koncentrációjának növekedése a különböző célsejtekben az erek simaizmai és a hörgők relaxációját, az energiatartalékok mozgósítását (a szénhidrátok lebomlását a májban), gátolja a vérlemezkék aggregálódási képességét, csökkenti a myometrium (méhizom) és a hólyag hangját stb.
Számos neurotranszmitter, például adrenalin (a β-adrenerg receptorok aktiválásával), dopamin (a D1-dopamin receptorok aktiválásával), az adenozin (az adenozin A2 receptorok aktiválásával) tartozik az endogén biológiailag aktív anyagokhoz, amelyek képesek aktiválni a G jelző fehérjéket. hisztamin (a hisztamin G 2 -receptor aktiválásával), szerotonin (a szerotonin 5-HT4-receptorok aktiválásával), valamint számos hormon, például vazopresszin (a V 2 -vasopressin receptorok stimulálása révén) stb.
Signal G i-proteinek. A G szignál fehérjékkel ellentétben a GI szignál fehérjék aktiválása nem stimulálja, hanem gátolja az effektor elem, az adenilát-cikláz enzim aktivitását, ami a cAMP koncentrációjának csökkenéséhez vezet a másodlagos hírvivő célsejtjeiben. A cAMP-tartalom csökkenése a célsejtekben a szív összehúzódásának csökkenését, az erek és a hörgők tónusának növekedését okozza, azaz a cAMP-tartalom növekedésének ellentétes hatása a célsejteken. Ezen felül számos jelző Gi fehérje részt vesz a transzmembrán ionos Ca 2+ és K + csatornák funkcionális aktivitásának szabályozásában.
Számos neurotranszmitter, például adrenalin és norepinefrin (egy 2-adrenoreceptor aktiválásával), dopamin (a D2-dopamin receptorok aktiválásával), az adenozin (az A1 aktiválásával) olyan endogén biológiailag aktív anyagokhoz tartozik, amelyek képesek aktiválni a G i-fehérjék jelét. adenozin receptorok), acetilkolin (az M 2 és M 4 muszkarin receptorok aktiválásával) stb.
Jel G ^ fehérjék. Ezek a jelző fehérjék hozzájárulnak a célsejtek másik effektor elemének, a foszforiláz C enzim aktiválásához, amely viszont serkenti a másodlagos hírvivők, a diacil-glicerin (DAG) és az inozitol-1,4,5-trifoszfát (ITP) képződését a célsejtekben. Ezek közül az első (DAG) kapcsolódik a sejtmembránhoz, és biokémiai reakciókat indít a kontraktilis állapot szabályozásában, a sejtnövekedésben és az osztódásban, valamint bizonyos hormonok célsejtek általi kiválasztásában. A foszfolipáz A 2 enzim hatására a DAG metabolizálódhat arachidonsavvá, amely részt vesz biológiailag aktív anyagok, például eikoszanoidok - prosztaglandini, prosztaciklinok, tromboxánok, leukotriének szintézisében (lásd T. I, 478. oldal).
A második szekunder messenger, az ITF, nem rögzül a sejtmembránon, és az intracelluláris közegbe (citoszolba) mozog, ahol elindítja a Ca2 + -ionok felszabadulását a sejt depókból, azaz elősegíti az inaktív Ca 2+ -ionok átalakulását az aktív formába.
Sok kutató úgy véli, hogy a Ca 2+ -ionok harmadlagos messenger vagy közvetítők. Ennek oka az a tény, hogy a Ca 2+ -ionok szerepe a sejtek funkcionális aktivitásának szabályozásában nagyon fontos. A Ca 2+ ionok a külső környezetből speciális transzmembrán ioncsatornákon keresztül juthatnak be a sejtekbe és / vagy a sejt depókból felszabadulhatnak. A fő raktár (az inaktív Ca 2+ -ionok felhalmozódásának helye) a sejtben az endoplazmás vagy szarkoplazmás retikulum (reticulum sarcoplasmaticunr, szinonimája: endoplazmatikus retikulum - intracelluláris organelle, amely a citoplazmában található tubulusok és ciszternák rendszere, amelyet a membrán korlátoz; anyagok szállítása a citoplazmában). A szarkoplazmatikus retikulumból a citoplazmába érkező szabad (aktív) Ca 2+ ionok kölcsönhatásba lépnek néhány Ca 2+ -kötő fehérjével, amelyek közül a legfontosabb a kalmodulin. A komplex "kalodulin-Ca 2+" és / vagy Ca 2+ -ionok komplexei más kalcium-kötő fehérjékkel kiváltják a sejtben zajló biokémiai reakciók sorrendjét. Ennek eredményeként a célszervektől függően, amelyben ez a folyamat zajlik, megindul a szívizom és a vázizom kontraktilis funkciójának fokozódása, az erek simaizomzatának, a hörgőknek és a méhnek a tónusa, fokozódik a mirigyszövetek szekréciós aktivitása, stimulálódik az idegvégződések idegátadók felszabadulása stb. . Azt is bebizonyították, hogy a Ca 2+ -ionok képesek növelni a fehérjék, szénhidrátok és zsírok metabolizmusában résztvevő enzimek aktivitását.
A szekunder hírvivők - DAG és ITF, tehát a szignál Gq fehérjék közötti közvetlen kapcsolaton túl - a Ca 2+ -ionok fiziológiás körülmények között meglehetősen összetett kölcsönhatásban vannak a cAMP szekunder hírvivővel, amelynek aktivitását a G jel és a G i fehérjék szabályozzák. Így kimutatták, hogy a szabad kalcium-ionok, amelyek egy idegsejt citoplazmájába lépnek be a kalodulin-Ca 2+ rendszeren keresztül, csökkenti az iAMP tartalmát a sejtben. Ugyanakkor a kalcium-ioncsatornák nyitott állapotának fenntartásához a sejtben magas cAMP-koncentrációra van szükség, azaz a cAMP-tartalom csökkenése, amelyet a kalmodulin-Ca 2+ komplex okozott, a szabad Ca 2+ ionok felvételének abbahagyását vonja maga után a citoplazmában. Másrészt bizonyítékok vannak arra, hogy a cAMP másodlagos hírvivő elősegíti a szabad Ca 2+ ionok abszorpcióját a szarkoplazmatikus retikulumban, azaz elősegíti a Ca 2+ -ionok átalakulását a szabad, aktív formáról a kötött, inaktív formára.
A célsejtekben - a DAG-ban és az ITF-ben - a másodlagos hírvivőanyag-tartalom növekedése eredményeként növekszik a simaizmok tonusa, növekszik a mirigyek szekréciója, elősegíti a neurotranszmitterek felszabadulását a preszinaptikus végződésekből, megkönnyíti a vérlemezkék aggregációs képességét stb.
Az endogén biológiailag aktív anyagok, amelyek képesek aktiválni a Cq szignálfehérjéket, ide tartoznak az olyan neurotranszmitterek, mint a norepinefrin (az 1-adrenoreceptorok aktiválásával), az acetilkolin (a muszkarin M1 és M3 receptorok aktiválásával), szerotonin (a szerotonin aktiválása miatt) 5-HT2a receptorok), hisztamin (a hisztamin H1 receptorok aktiválása miatt), valamint más endogén biológiailag aktív anyagok, például bradykinin és angiotenzin.
Jelenleg a felsorolt \u200b\u200bszignál G-fehérjék (Gs, G |, Gq) mellett más G-szignál-fehérjéket is azonosítottak - Gs, Gi, Gq, amelyek fiziológiai szerepe továbbra sem teljesen tisztázott. Ugyanakkor bizonyítékok vannak arra, hogy például a C o jelű protein részt vesz a transzmembrán ioncsatornák funkcionális aktivitásának szabályozásában.
A 11. típusú receptorok funkcionális egysége egy transzmembrán (a sejtmembrán teljes vastagságát áthatoló) fehérje (enzim). Maga a receptor két azonos fragmensből áll, amelyeket monomereknek hívnak. A monomerek jelentéktelen távolságra vannak egymástól, és maga a monomer két funkcionálisan aktív alegységből - doménből áll, amelyeket egy polipeptid szegmens köt össze, amely keresztezi a lipid kettős réteg membránját (1.9. Ábra). A monomer a-alegysége kinyúlik a membrán külső felülete felett, és felelős a receptor biológiailag aktív anyagokhoz történő kötődéséért, és a P-alegység a sejt citoplazmájába merül.

Ábra. 1.9. A II. Típusú receptor szerkezete (magyarázat a szövegben): 1 - a monomer a-alegysége; A monomer 2 - β-alegysége
A biológiailag aktív anyagnak a receptor α-alegységéhez történő kötődése után a receptor inaktív monomer állapotból aktív dimer állapotba változik, amelyben két monomer a membrán síkjában egyesül (lásd 1.9 ábra). Ebben az esetben stimulálódik a receptor citoplazmatikus β-alegységének enzimatikus aktivitása, amelynek eredményeként a célsejtben elindul a funkcionális állapotát megváltoztató biokémiai reakciók kaszkádja.
A receptort képező transzmembrán enzimként általában enzimeket, például tirozin-kinázt vagy guanilát-ciklázt alkalmazunk.
A tirozin-kináz-receptor példája az inzulinreceptorok (lásd T. 1, 435. oldal).
A guanilát-cikláz szignál átviteli útja a receptor α-alegységének kölcsönhatásba lép egy endogén biológiailag aktív anyaggal, például pitvari natriuretic faktorral (ANF), amely biológiailag aktív anyag, amelyet pitvari sejtek választanak ki és részt vesznek a szív összehúzódások szabályozásában. Ezen kölcsönhatás eredményeként megváltozik a receptor konfigurációja, amely monomereinek dimerré történő kombinálását foglalja magában. Ez a folyamat aktiválja a receptor enzimrészét, amely a citoszolos P-alegységében található, azaz a guanilát-cikláz enzim, amely elősegíti a ciklikus guanidin-3,5 "monofoszfát (cGMP) másodlagos hírvivőanyag koncentrációjának növekedését a célsejtben. A cGMP koncentrációjának növekedése a célsejtekben olyan biokémiai reakciók sorozatát váltja ki, amelyek megváltoztatják funkcionális állapotukat, például a simaizomsejtek relaxációját hajó.
A III. Típusú receptorok olyan receptorokat tartalmaznak, amelyek endogén biológiailag aktív anyagok - neurotranszmitterek - befolyása alatt biztosítják a megfelelő ionok átjutását a sejtmembránon, ami megváltoztatja annak (membrán) elektromos töltését (potenciálját).
Szerkezetükben a III tina receptorok olyan csatornát képviselnek, amely áthatol a sejtmembrán lipid kettős rétegében, és több polipipes egység alkotja (1.10. Ábra). Például a nikotin (H) receptor egy 8 nm átmérőjű csatorna, amelyet öt polipeptid alegység (a - kettő, β, γ, d) alkot (lásd 1.10. Ábra). Amikor az acetilkolin neurotranszmitter kölcsönhatásba lép a sejtmembrán felszíne felett kinyúló résszel (doméntel) - a receptor α alegységével -, szerkezete megváltozik, és egy központi csatorna nyílik, amelyen keresztül a Na + -ionok a koncentrációgradiens szerint belépnek a célsejtbe, ami megváltoztatja annak funkcionális tulajdonságait. aktivitást. A H-kolinerg receptorokon kívül a gamma-amino-vajsav és az izgató aminosavak receptorai is a III. Típusú receptorokhoz tartoznak.
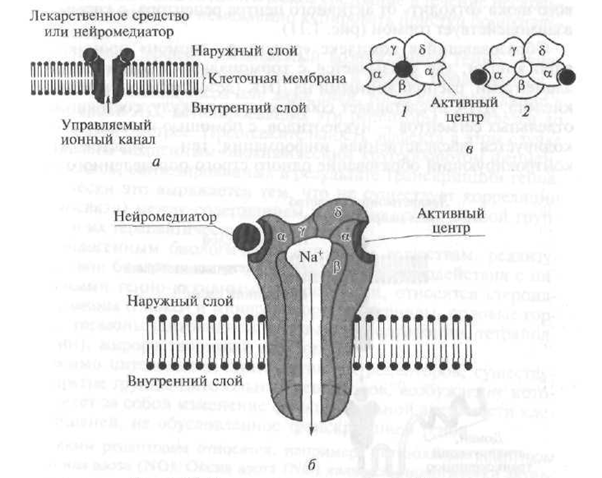
Ábra. 1.10. A 111 típusú receptor szerkezeti diagramja:
a egy kapcsolási rajz; b - transzmembrán ioncsatorna (összefüggésben); c - transzmembrán ioncsatorna (felülnézet); / - csatorna inaktív (zárt) állapotban; 2 - csatorna aktív (nyitott) állapotban; a. β, γ, d - csatornás polipeptid alegységek
A IV. Típusú receptorok közé tartoznak az intracelluláris és a nukleáris receptorok. Az ilyen típusú receptorokkal kölcsönhatásba lépő biológiailag aktív anyagok lipofil (zsírokban könnyen oldódó) vegyületek, ezért könnyen behatolnak a sejtmembránba, és elérik az intracelluláris receptorokat. Az intracelluláris receptorok magukban foglalják a hormonok receptorait, valamint más biológiailag aktív anyagokat.
A hormonok és az intracelluláris receptorok kölcsönhatásának mechanizmusa meglehetősen bonyolult, azonban vázlatosan a következőképpen ábrázolható. Szerkezetük szerint a hormonok intracelluláris receptorja egy polipeptid, amely több funkcionális egységből - doménből áll. Hormon hiányában a receptor inaktív, mivel aktív központját egy speciális fehérje - az úgynevezett hő-sokk protein - blokkolja. Abban az esetben, ha a hormon „megközelíti” a receptort, akkor a hőgumi fehérje „eltér” a receptor aktív központjától, amellyel a hormon kölcsönhatásba lép (1.11. Ábra).
A kapott receptor-hormon komplex áthatol a sejtmagjában, ahol a DNS-en található dezoxiribonukleinsavhoz kötődik hormonérzékeny elemekhez (dezoxiribonukleinsav; a DNS egy makromolekulája, amely külön szegmensekből áll - nukleotidok, amelyek segítségével az öröklött információ a génekbe van kódolva; a gén - egy DNS-darab, amely szabályozza egy szigorúan meghatározott fehérje képződését
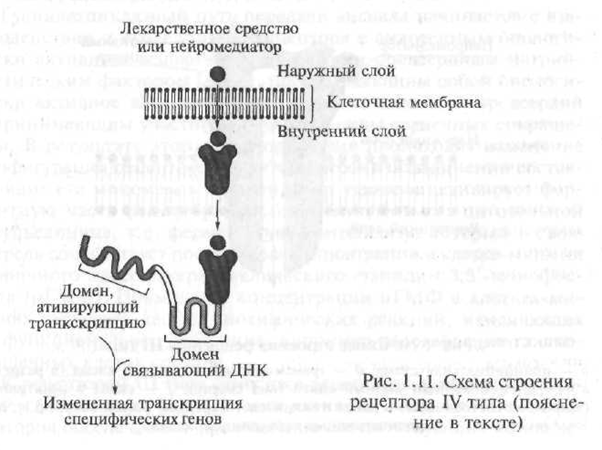
Ábra. 1.11. A IV típusú receptor szerkezetének vázlata (magyarázat a szövegben)
ka). Ennek a kölcsönhatásnak a eredményeként megkezdődik a génátírási folyamat - a genetikai kódban levő információnak a DNS-molekuláról az információs RNS (mRNS, syn: mátrix RNS - mRNS) molekulájára történő átvitele. A transzkripció az első lépés a fehérjék képzésében egy sejtben. A kapott mRNS. elhagyja a sejtmagot, és a riboszómákba mozog - az intracelluláris organellák, amelyek felelősek a fehérje szintézisében a sejtben. A speciális orvosi szakirodalomban azokat a receptorokat, amelyek aktiválása okozza a gén transzkripcióját, genetikailag aktív receptoroknak nevezik.
Általában a célsejtek reakciója a gén-aktív receptorok gerjesztésére lassan alakul ki, ami klinikai szempontból nagyon fontos.
Először, a célsejtek válaszideje késik az időben, mivel ehhez új fehérjék szintézisére van szükség, amely általában 20-30 percet vesz igénybe, azaz A hormonok, amelyek aktiválják a IV. típusú receptorokat, néhány perc alatt nem képesek megváltoztatni a kóros állapotot, például azonnal megállítják a hörgőasztma rohamait.
Másodszor, a gén-aktív receptorok gerjesztése által okozott hatás meglehetősen hosszú, és akár több órát vagy akár napot is eltarthat, miközben ezeket a receptorokat aktiváló gyógyszerek vérplazma-tartalma sokkal gyorsabban nullára csökken. A hatás időtartama ebben az esetben a géntranszkripció eredményeként szintetizált enzimek és fehérjék lassú biokémiai keringésének köszönhető. Klinikailag ezt az a tény fejezi ki, hogy nincs kapcsolat (kapcsolat) egy adott gyógyszercsoport plazmatartalma és terápiás hatása között.
Az endogén biológiailag aktív anyagok, amelyek biológiai hatásaikat a citoszol gén-aktív receptorokkal való kölcsönhatás révén realizálják, a szteroid hormonok (gluko- és mineralokortikoszteroidok, nemi hormonok), pajzsmirigyhormonok (triiodothyronine, tetraiodothyronine) és zsírban oldódó D-vitamin.
A citoszolos gén-aktív receptorokon kívül a citoszolos receptorok más csoportjai is vannak, amelyek gerjesztése a célsejtek funkcionális aktivitásának megváltoztatását vonja maga után, nem a gének transzkripciója miatt.
Ilyen receptorok például a salétrom-oxid (N0) citoszolos receptorai. A salétrom-oxid (N0) biológiailag aktív anyag, amely az érrendszeri endotéliumban képződik. Endogén biológiailag aktív anyagként a salétrom-oxidot először az amerikai fiziológus R. F. Furchgott 1987-ben izolálta a nyulak kikötőiből, és "endothelial relaxing factor - ORF" -nek hívták. A salétrom-oxid egy lipofil vegyület, amely könnyen behatol a sejtmembránba, ahol kölcsönhatásba lép specifikus citoszol receptorokkal, ami magában foglalja a guanilát-cikláz enzim aktiválását. Ez utóbbi viszont serkenti a cGMP szekunder hírvivő szintézisét, amely kaszkádban indítja az intracelluláris biokémiai reakciókat, amelyek a célsejtek - érrendszeri simaizomsejtek - relaxációjához vezetnek.
Ennélfogva jelenleg négy fő mechanizmust és ennek megfelelően az IV receptor típusokat különböztetjük meg annak kölcsönhatása miatt, amelykel az endogén biológiailag aktív anyagok és / vagy azok szintetikus analógjai, azaz A gyógyszerek befolyásolhatják a célsejtek funkcionális állapotát.
Ez azonban nem azt jelenti, hogy a biológiailag aktív anyagok ismert receptorainak száma 4-re korlátozódik. Ezek mérhetetlenül nagyobbak. Ennek oka az a tény, hogy ugyanazon alapvető hatásmechanizmuson keresztül nagyon sok különböző kémiai szerkezetű endogén biológiai anyag befolyásolhatja a sejtek funkcionális aktivitását. Például a norepinefrin és a hisztamin neurotranszmitterek, amelyek kémiai szerkezetükben különböznek, és ezért azokban a receptorokban, amelyekkel kölcsönhatásba lépnek, gerjesztő jelet továbbítanak a célsejtekhez ugyanazon alapvető mechanizmus révén - a G szignál fehérjék aktivitásának stimulálása, azaz mindkettő kölcsönhatásba lép az I. típusú receptorokkal.
Ezért az összes jelenleg ismert receptort nemcsak a célsejtek intracelluláris struktúrájába történő jelátvitel tulajdonságai alapján osztályozzuk, hanem azon endogén biológiailag aktív anyagok nevei alapján is, amelyekkel specifikusan kölcsönhatásba lépnek.
Meg kell jegyezni, hogy a receptorok megkapták a nevüket, figyelembe véve az endogén biológiailag aktív anyagok nevét, amellyel kölcsönhatásba lépnek, jóval azelőtt, hogy a célsejtekbe történő jelátvitel mechanizmusai ismertté váltak.
Az endogén biológiailag aktív anyagok, amelyek a specifikus receptorokkal való kölcsönhatás révén realizálják hatásaikat, ide tartoznak a neurotranszmitterek (acetilkolin, norepinefrin, dopamin, hisztamin, szerotonin stb.), Hormonok, szöveti eredetű biológiailag aktív anyagok - autocaidok (prosztaglandinok, tromboxánok, leukotriének, bradykinin, angiotenzin stb.). A speciális orvosi szakirodalomban ezeket az anyagokat gyakran kombinálják „ligandumok” kifejezéssel (a Lat. Ligo-tól - kötődnek, vagyis egy olyan anyagot, amely képes a receptorhoz kötődni).
Így a receptorok a specifikus ligandumaik neve alapján kapják a nevüket. Például a dopamin neurotranszmitter receptorait dopaminnak, inzulinhormonnak - inzulint, leukotrién augocaidot - leukotriént stb. Nevezzük.
A jövőben a tankönyv szövegében azért, hogy elkerüljük a zavart a receptorról az intracelluláris képződményekre történő jelátvitel mechanizmusában, a „receptor típusa” kifejezést használjuk, és ha a receptor nevéről beszélünk, mivel a ligandum kölcsönhatásba lép vele. , a „receptorfaj” kifejezést kell használni.
Általános szabály, hogy ugyanazon faj sok receptorát több altípusra osztják, például az adrenerg receptorokat a- és β-adrenerg receptorokra, a kolinerg receptorokat M- és N-kolinerg receptorokra osztják stb. A legtöbb esetben az alfajokat kisebb csoportokra is fel kell osztani: β 1 - és β 2 -adrenoreceptorok, N n - és N m-kolinerg receptorok stb.
A modern farmakológia szempontjából nagyon fontos a receptorok alfajainak azonosítása és az endogén biológiailag aktív anyagok kölcsönhatásba lépésének mechanizmusainak tanulmányozása, mivel lehetővé teszi olyan gyógyszerek létrehozását, amelyek kölcsönhatásba lépnek a receptor szigorúan meghatározott alfajaival. Tehát például a β-adrenerg receptorok megoszlása \u200b\u200bβ 1-re (elsősorban a szívsejtek sejtmembránján) és β 2 -re (lokalizálva például a hörgők simaizomsejtjeinek membránjain) lehetővé tette olyan gyógyszerek létrehozását, amelyek szelektíven hatnak a szívizomra (β 1 -adrenostimulánsok). ) - nonachlazin gyógyszer, és szelektíven befolyásolja a hörgők simaizmait (β 2 -adrenostimulánsok) - salbutamol gyógyszer stb.
Meg kell jegyezni, hogy a test szervei és szövetei nem tartalmaznak állandó számú receptort és / vagy alfaját, azaz változó. Mind a kóros folyamatok, mind a gyógyszerek megváltoztathatják a szervek receptorainak számát.
Például a szívkoszorúér betegséggel növekszik a szívizomban a 3-adrenerg receptorok száma, magas vérnyomásban szenvedő betegeknél pedig az a- és a β-adrenerg receptorok száma is növekszik.Az antidepresszáns imipramin hosszú távú használata csökkenti a β-adrenerg receptorok mennyiségét az agyszövetben. Igen sok ilyen példa létezik.
Az endogén (testben termelődött) neurotranszmitterek vagy gyógyszerek affinitását a receptorokhoz az „affinitás” kifejezés jellemzi, és a receptorokhoz való kötődésük sebességét és erősségét az „affinitás” kifejezés jelöli.
A gyógyszerek kölcsönhatása a receptorral természetesen nem öncél, hanem változásokhoz vezet a szervek vagy a test szöveteinek aktivitásában.
Az ilyen változást vagy reakciót, amely megfelel ennek a receptornak a funkcionális jelentőségére, a gyógyszerek belső aktivitásának nevezzük.
Az intrinsic aktivitással és receptor affinitással rendelkező gyógyszerek agonisták, azaz úgy viselkednek, mint endogén biológiailag aktív anyagok.
Például a fenilefrin, az a-adrenerg receptorok stimulátora, olyan hatást gyakorol az arteriolákra, mint a norepinefrin neurotranszmitter, azaz az a-adrenerg receptorok agonistája. A speciális orvosi szakirodalomban az „agonista” kifejezésen túlmenően a „receptor stimuláns” vagy „utánzó” kifejezést is használják, például egy adrenomimetikum, azaz Az adrenerg receptorokat stimuláló gyógyszerek.
Antagonistáknak nevezzük azokat a gyógyszereket, amelyek receptor affinitással rendelkeznek, de gátolják az exogén és endogén agonisták kölcsönhatását a receptorral.
Például az atropin M-kolinerg receptor blokkoló zavarja az acetilkolin neurotranszmitter M-kolinerg receptor kölcsönhatást; blokkoló (propranolol J-adrenerg receptorok, amelyek blokkolják a tüdő β 2 -adrenostruktúráit) gátolja a szalbutamol β 2 -adrenostimulátor stimuláló hatását rájuk, azaz az atropin és a propranolol a megfelelő receptorok antagonistái.
A szakorvosi szakirodalomban az „antagonista” kifejezésen túl a „receptor blokkolók” vagy „lítik” kifejezés, például antikolinerg szerek, azaz a kolinerg receptorokat gátló gyógyszerek.
A receptorok kölcsönhatását az agonistákkal és antagonistákkal vázlatosan szemléltetve a 2. ábrán mutatjuk be. 1.12.
Az agonisták mind közvetlen, mind közvetett hatást gyakorolhatnak, azaz közvetett cselekvés.

Ábra. 1.12. A receptor és az agonista (a) és az antagonista (6) és a receptor kölcsönhatásának vázlata (magyarázat a szövegben)
Például egy opioid receptor agonista - a morfin gyógyszer - a (mu) p-, (kappa) k- és (delta) 8-opioid receptorok közvetlen stimulálásával valósítja meg hatásait, azaz közvetlen stimuláló hatást gyakorol rájuk, míg a szimpatomimetikus efedrin sejtes hatásait az a- és β-adrenerg receptorok közvetett vagy közvetett stimulálásával valósítja meg: segít kiszorítani a norepinefrin neurotranszmitter preszinaptikus idegvégződéseit, gátolja az idegvégződések általi visszavételét, és növeli az a- és p- adrenoreceptorokat ad a norepinefrinhez és epinefrinhez, és serkenti az adrenalin felszabadulását a mellékvesekéregből. Így az efedrin farmakológiai hatásait nem közvetlenül, hanem egy neurotranszmitter révén valósítja meg, miközben nincs közvetlen interakcióban a receptorral.
Az antagonisták, valamint a receptor agonisták farmakológiai hatásaikat közvetlenül vagy közvetetten észlelhetik, blokkolva a megfelelő receptorokat.
Például a gyomor H3 hisztamin receptor blokkolója, a ranitidin készítmény közvetlenül kölcsönhatásba lép a gyomor H 2 hisztamin receptorokkal, gátolja a sósav szekrécióját, gátolja az interakciót az endogén hisztamin neurotranszmitter receptorral, azaz közvetlenül blokkolja a receptort, miközben a testbe belépő szimpatolitikus rezerpin, mint a szimpatomimistás efedrin, „kiüríti” a norepinefrin neurotranszmitterét a preszinaptikus végződésekből, miközben ezzel egyidejűleg blokkolja a norepinefrin új „adagjainak” szintézisét, ami a mediátorkészletek gyors elfogyásához vezet a presynapticus végén. Ennek eredményeként állapot alakul ki, amikor az erek a-adrenerg receptorai "nem működnek" fiziológiai stimulátor noradrenapin hiánya miatt. Azt mondhatjuk, hogy a szimpatolitikus rezerpin az a-adrenerg receptorok funkcionális, közvetett blokádját okozza.
Egyes gyógyszerek egyesítik az agonisták és antagonisták tulajdonságait, azaz bizonyos körülmények között ugyanazok a receptorok gerjesztik vagy blokkolják. Azokban az esetekben, amikor a stimuláló komponens a gyógyszer farmakológiai hatásában uralkodik, azt mondják, hogy a gyógyszer részleges vagy részleges agonista.
A parciális vagy részleges agonisták hatásmechanizmusa az, hogy ezek a gyógyszerek, megközelítve a receptort, rajta vannak rögzítve. Kémiai szerkezetük sajátosságai miatt ugyanakkor gerjesztik, hogy nem teljes, hanem csak részleges stimuláló választ okoznak. Ugyanakkor zavarják más gyógyszerek és / vagy a megfelelő neurotranszmitter kölcsönhatását e receptorral.
Például a nalorfin, amelyet narkotikus fájdalomcsillapítók túladagolásának kezelésére használnak, részleges agonista. A Nalorfin, amely "felmegy" az opioid receptorhoz, kiszorítja a morfint a kapcsolatából, vagyis megállítja a morfin agonista hatását a receptorra. Ha a nalorfin a receptorral érintkezik, akkor kis mértékben stimulálja azt, amely nem képes elnyomni a légzőközpont aktivitását, azaz részleges agonista hatással van az opioidreceptorra, ezzel párhuzamosan blokkolja azt a morfin számára, ezáltal antagonista hatást mutatva a morfinra nézve. A nalorfin hátránya, hogy túladagolás esetén fokozza a receptorra gyakorolt \u200b\u200bstimuláló hatását, gátolja a légzési központot, valamint a morfint.
Azokban az esetekben, amikor a gyógyszerek farmakológiai hatásában blokkoló hatás dominál, azt mondják, hogy a gyógyszer antagonista, saját vagy belső aktivitásával.
Az antagonisták saját aktivitással bíró hatásmechanizmusának sajátosságai az acebutalol β 1 -adrenoblokkolójának példájában mérlegelhetők. Az acebutalol a β 1 \u200b\u200b-adrenerg blokkolók csoportjába tartozik, saját belső szimpatomimetikus aktivitással, azaz egyesíti a β 1 \u200b\u200b-adrenoreceptorok blokkoló (lítikus) és stimuláló (mimetikus) tulajdonságait.
Az acebutalol hatásmechanizmusa annak kémiai szerkezetének sajátosságaiból fakad, amelynek következtében a gyógyszer egyrészt blokkolja a szívizom β 1 -adrenoreceptorokat, másrészt serkenti azok aktivitását, ezáltal szimulálva a mediátorok - katecholaminok fiziológiás hatását (lásd T. 1. p. 170).
Valamennyi gyógyszert a hatásuk sajátosságai alapján két nagy csoportra lehet osztani - specifikus és nem-specifikus hatásokkal.
A nem-specifikus hatású gyógyszerek közé tartoznak azok a gyógyszerek, amelyek széles skálájú farmakológiai aktivitással rendelkeznek, és a hatások különböző alkalmazási pontjai. Ebbe a csoportba tartoznak a vitaminok, biogenikus stimulánsok, antioxidánsok, adaptogének stb.
A specifikus hatású gyógyszerek közé tartoznak azok a gyógyszerek, amelyeket a megfelelő receptorok agonistáinak vagy antagonistáinak tulajdonságai jellemeznek, azaz hatásmechanizmusuk alapja az a képesség, hogy kölcsönhatásba lépjenek számukra specifikus receptorokkal.
Meg kell azonban jegyezni, hogy az olyan gyógyszerek farmakológiai hatása, amelyeknek specifikus hatása van bármely receptorra, nem mindig lesz nagyon specializálódott, például csökkentheti a pulzusszámot (HR) vagy elnyomja a gyomornedv szekrécióját. Számos gyógyszer teljes farmakológiai hatása attól függ, hogy hány és milyen szervekben és szövetekben vannak a gyógyszerre specifikus receptor képződmények.
Például a β-adrenerg receptorok a szívizomban, érrendszeri falban, hörgőkben, méhben, zsírszövetben, csontvázizomban stb. Találhatók. Ennek eredményeként a p-adrenerg receptorokat stimuláló gyógyszerek bizonyos mértékig növelik a szív összehúzódásainak erősségét és gyakoriságát, az erek tágulását, a hörgőt és a méh alsó tónusát, azaz szisztémás hatást gyakorolnak a testre.
A gyógyszerek specifikus hatása lehet szelektív vagy szelektív, és ennek megfelelően nem szelektív vagy nem szelektív. A gyógyszerek szelektivitását az határozza meg, hogy befolyásolja-e bármely receptor összes alfaját vagy azok specifikus alfajait.
Például a nem szelektív β-adrenerg blokkoló propranolol blokkolja mind a szívizomszövetben elhelyezkedő β 1 -adrenoreceptorokat, mind pedig a tüdőszövetben elhelyezkedő β 2 -adrenoreceptorokat, azaz nem-szelektív (nem szelektív) hatással van a β-adrenerg receptorok mindkét alfajára. . mivel a szelektív β 1 -adrenerg blokkoló atenolol terápiás adagokban csak a β 1 \u200b\u200bszívizom-adrenerg receptorokat gátolja, azaz szelektív (szelektív) hatást fejt ki egy specifikus β-adrenerg receptorra.
A receptor alfajok fiziológiai szerepének és lokalizációjának tisztázása lehetővé teszi olyan nagyon hatékony gyógyszerek létrehozását, amelyek szelektíven hatnak a receptorok különböző alfajaira.
Például a β 1 \u200b\u200b-adrenerg receptorokat gátló gyógyszereket széles körben használják a szívkoszorúér betegség, artériás hipertónia és a β 2 -adrenerg receptorokat stimuláló gyógyszerek alkalmazásában a hörgő asztma kezelésére. A H1 hisztamin receptorokat gátló gyógyszereket lehetőleg a klinikai gyakorlatban írják elő a megelőzés és / vagy letartóztatás érdekében allergiás reakciókmíg a H2 hisztamin receptorokat gátló gyógyszerek hatékonyan kezelik a gyomor- és nyombélfekélyeket.
A szelektív hatású gyógyszerek előállítása nagymértékben optimalizálta klinikai felhasználásukat, mivel jelentősen csökkent mellékhatás.
A gyógyszerek hatásmechanizmusának alapja általában az, hogy képesek olyan komplex biokémiai és / vagy biofizikai folyamatokat kezdeményezni (kiváltani), amelyek végül megváltoztatják és / vagy optimalizálják a célsejt funkcionális aktivitását. A gyógyszerek megismerhetik a szervekre és / vagy vagy célsejtek: közvetlen kémiai kölcsönhatás révén; fizikai-kémiai kölcsönhatás a sejtmembránon; a speciális enzimekre gyakorolt \u200b\u200bhatások; a szabályozó génekre gyakorolt \u200b\u200bintézkedések; a specifikus receptorokra gyakorolt \u200b\u200bhatások.
A drogok közvetlen kémiai kölcsönhatása.
A gyógyszerek ezen hatásmechanizmusa meglehetősen ritka, és a sejtön kívül is megvalósítható, például a gyomor vagy a bél lumenében. Lényege abban rejlik, hogy a gyógyszerek közvetlen kémiai reakcióba lépnek olyan molekulákkal és / vagy ionokkal, amelyek a testben normál vagy kóros állapotban képződnek. A közvetlen kémiai kölcsönhatás példája a gyomor sósavjának semlegesítésének kémiai reakciója savmegkötő gyógyszerek szedésekor.
A gyógyszerek fizikai-kémiai kölcsönhatása a sejtmembránon.
A citoplazmatikus membrán egyik fő funkciója az ioncserék megvalósítása a citoplazma és az extracelluláris környezet között. A transzmembrán ioncserére speciális feszültségfüggő transzmembrán ioncsatornákon keresztül is sor kerülhet - nátrium, kálium, kalcium, klór stb. Egyes gyógyszerek, elérve a sejtmembránt, kölcsönhatásba lépnek ezekkel a csatornákkal és megváltoztatják funkcionális aktivitásukat. Tehát például az IA osztályú gyógyszer - a kinidin - antiaritmiás hatása azon a képességén alapszik, hogy megakadályozza a Na + -ionok átjutását a transzmembrán nátrium-csatornákon keresztül.
A gyógyszerek hatása a speciális enzimekre.
Egy viszonylag kis mennyiségű gyógyszer realizálja farmakológiai hatását, amikor megváltoztatja egyes specializált sejtes enzimek aktivitását. A celluláris enzimek aktivitását fokozó gyógyszereket nevezik induktorokenzimeket. Ilyen hatást gyakorolnak például altatók és görcsoldó fenobarbitál gyógyszerek, amelyek jelentősen növelik a mikroszomális májenzimek aktivitását. A fenobarbital és rokon gyógyszerek ezen hatásának biológiai jelentőségét az alábbiakban tárgyaljuk.
Gyógyszereket hívnak, amelyek gátolják a speciális enzimek aktivitását inhibitorokenzimeket. Tehát például a monoamin-oxidáz-gátlók (MAO) csoportjából származó antidepresszánsok esetében a pirlindol gyógyszer az antidepresszáns hatását úgy realizálja, hogy gátolja a MAO enzim aktivitását a központi idegrendszerben. Az acetilkolinészteráz enzim aktivitásának gátlására való képesség alapját képezik az antikolineszteráz gyógyszerek, például a fizosztigmin farmakológiai aktivitása. Ismeretes, hogy fiziológiás körülmények között az acetilkolinészteráz inaktiválja (elpusztítja) az acetilkolint, egy neurotranszmittert, amely gerjesztést közvetít a parasimpatikus idegrendszer szinapszisában. A fizosztigmin, az acetilkolinészteráz aktivitását elnyomva, elősegíti az acetilkolin neurotranszmitterének parasimpátikus rendszerének szinapsáiban történő felhalmozódást, amelynek eredményeként a parasimpatikus idegrendszer tónusa megemelkedik, ami szisztémás szinten a bradycardia kialakulásával (növekvő vérnyomás, alacsonyabb szintű vérnyomás, BP) növekszik. tanuló stb. A gyógyszerek visszafordíthatóan és visszafordíthatatlanul kölcsönhatásba léphetnek az enzimekkel. Például az enalapril gyógyszer visszafordíthatóan gátolja az angiotenzin-konvertáló enzim aktivitását, ami különösen a vérnyomás csökkenését vonja maga után, míg a szerves foszfor-mérgező anyagok visszafordíthatatlanul gátolják az acetilkolinészteráz aktivitását. A gyógyszerek hatása a szabályozó génekre.Jelenleg a tudósok olyan gyógyszereket próbálnak kidolgozni, amelyek felismerik farmakológiai hatásaikat azáltal, hogy közvetlenül befolyásolják a szabályozó gének fiziológiai aktivitását. Ez a tendencia különösen ígéretesnek tűnik azután, hogy az emberi genom szerkezetét 2000-ben megfejtették. Úgy gondolják, hogy a szabályozó gének működésének szelektív normalizálása gyógyszerek hatására lehetővé teszi számos betegség, beleértve a korábban gyógyíthatatlan betegségek kezelését is. A gyógyszerek hatása a receptorokra.Mielőtt továbblépnénk a gyógyszerek és a receptorok közötti kölcsönhatás jellemzőire, tisztáznunk kell, hogy mit értünk a „receptor” kifejezés alatt (lat. recipio - vegye, vegye). A fiziológia kezdetéből ismert, hogy a "receptor" kifejezés olyan speciális képződményeket jelent, amelyek képesek érzékelni, átalakítani és továbbítani a külső jel energiáját az idegrendszerbe. Ezeket a receptorokat nevezzük szenzoros(a lat. sensus - érzés, érzés, érzékelés). Az érzékszervi receptorok magukban foglalják a hallás, látás, illat, íz, érzés, szervek receptorait. Ezen szervek szenzoros receptorai az úgynevezett exteroreceptorokhoz tartoznak. Ha az ősi idők óta ismertek azok a szenzoros szervek, amelyek reagálnak a külső irritáció stimulusokra, akkor a testben található szenzoros receptorok jelenlétét a 19. század közepéig megkérdőjelezték. Az első orosz fiziológus I. I. Zion, amely 1866-ban nyúlkísérlet során az aorta irritációja miatt csökkentette a vérnyomást. Ez a felfedezés a test belsejében levő receptorok kutatására és tanulmányozására vezetett, és magukat ezeket a receptorokat nevezték el interoreceptorokhoz. Kszázad eleje elegendő számú szenzoros interoreceptort fedeztünk fel, és bizonyították azok fontos szerepét a test élettani funkcióinak szabályozásában. 1905-ben J. Langley bebizonyította, hogy amikor egy gyógyszert feltesznek egy sejtmembránra, akkor farmakológiai hatás alakul ki, ha csak annak egy meghatározott részére alkalmazzák. Ezenkívül ez a hely a sejtfelület teljes területének csak kis részét teszi ki. Ez a megfigyelés lehetővé tette J. Langley arra a következtetésére, hogy a gyógyszerekkel kölcsönhatásba lépő speciális receptor helyek léteznek a sejtmembránon. A drogok hatásának receptor elméletének megalkotásában azonban a prioritás a német élettárs, P. Ehrlich, aki 1906-ban először vezette be a „receptor” kifejezést, és megfogalmazta azt az állítást, hogy „a gyógyszer nem működik, ha nem rögzítve van a sejtmembránon”. P. Ehrlich elmélete szerint a gyógyszermolekulának két funkcionálisan aktív csoportja van, amelyek egyike biztosítja a rögzítését a sejtfelszínen a gyógyszerreceptor régiójában, a második funkcionális csoport kölcsönhatásba lép a receptorral, és olyan biokémiai reakciók komplex láncát indítja el, amelyek megváltoztatják annak (sejt) fiziológiai aktivitását. . Így már a 20. század elején. nyilvánvalóvá vált, hogy az interoreceptoroknak legalább két osztálya van: szenzoros receptorokinformáció továbbítása a központi szervek és a test szöveteinek állapotáról a központi idegrendszerben; sejtreceptorokamelyek kölcsönhatásba lépnek olyan gyógyszerekkel, amelyek megváltoztatják a célsejtek funkcionális aktivitását.<>Azonnal meg kell jegyezni, hogy a jövőben a tankönyv szövegében a terminológiában, a gyógyszerreceptorokban és a biológiailag aktív anyagokban, azaz a zavarok elkerülése érdekében, azaz sejtes vagy cytoreceptory,a „receptor” kifejezéssel, míg az érzékszervi interoreceptorokat funkcionális aktivitásukat jellemző kifejezéssel jelölik, például „baroreceptors”, „fájdalomreceptorok” stb. P. Ehrlich felfedezése a gyógyszerreceptorok sejtmembránján alapul szolgált a farmakológiai tudomány, különösen a farmakodinamika fejlődésében, amelynek egyik fő feladata a gyógyszerek receptor-hatásmechanizmusainak tanulmányozása. Jelenleg számos sejtreceptor felépítését, egyes biológiailag aktív vegyületekkel való kölcsönhatásuk tulajdonságait tárják fel, amelyek egyrészt lehetővé tették az ismert gyógyszerek hatásmechanizmusának megértését, másrészt pedig az alapját új, nagyon hatékony gyógyszerek létrehozásához. Természetesen nehéz elképzelni, hogy az evolúció során különböző szintetikus (kémiai úton előállított) gyógyszerek receptorai alakulnak ki az emberi testben, különösen mivel a modern gyógyszerpiacon bemutatott gyógyszerek túlnyomó többségét az elmúlt 50 évben vagy annál kevesebbel állították elő. Bebizonyosodott, hogy a sejt receptor berendezése egy nagyon ősi funkcionális-szerkezeti formáció. Tehát az a- és b-adrenoreceptorokat (amelyek olyan receptorai, amelyeknek a norepinefrin és az adrenalin kölcsönhatása befolyásolja a sejt funkcionális aktivitását) nemcsak az állati sejtekben, hanem a növényi sejtek membránjain is megtalálják, például a növényi nittella sejtjeiben, ahol a- és b- adrenoreceptorok szabályozzák a protoplazma (sejttartalom) mozgását. Akkor mi a P. Ehrlich által felfedezett drogok receptora, és miért lépnek kölcsönhatásba velük? Jelenleg nem kétséges, hogy az úgynevezett gyógyszerreceptorok valójában endogén (a szervezetben előállított) biológiailag aktív anyagok receptorai, amelyek részt vesznek a belső szervek és a test szövetek funkcionális aktivitásának szabályozásában. Az ilyen biológiailag aktív vegyületek közé tartoznak az idegvégződésekből az idegjel továbbításakor felszabadult anyagok, valamint a hormonok, vitaminok, aminosavak stb. Minden endogén biológiailag aktív anyaghoz szigorúan specifikus receptor van. Tehát például a testben előállított biológiailag aktív anyag, az adrenalin képes szigorúan specifikus a - és b-adrenerg receptorok, valamint a glükokortikoszteroidok - a mellékvesekéreg hormonjai - csak a szigorúan rájuk specifikus glükokortikoszteroid receptorokkal lépnek kölcsönhatásba. Azok a szintetikus gyógyszerek, amelyek a sejtek receptúrájával kölcsönhatásba lépve, kémiai szerkezetükben észlelik, többé-kevésbé hasonlítanak az endogén biológiailag aktív vegyületekhez, amelyek kölcsönhatásba lépnek hasonló receptorokkal. Például, a szintetikus vazokonstriktorok (vazokonstrikciót okozó) gyógyszerek, a fenilefrin kémiai szerkezete közel áll az endogén biológiailag aktív anyag norepinefrinhez, ezért, mint a norepinefrin, képes stimulálni az adrenoreceptorokat. Időnként kémiai szerkezetük sajátosságai miatt a gyógyszerek kölcsönhatásba léphetnek nem magával a receptorral, hanem a sejtmembrán szomszédos részével. Mivel ebben az esetben a gyógyszer nem magával a receptorral, hanem a sejtmembrán szomszédos részével lép kölcsönhatásba, nem izgalmas vagy blokkoló hatást fejtenek ki a receptorra, hanem allostemetrikus(görögül allos - másik, eltérő hatás vagy hatás. Ennek eredményeként megváltozhat mind a receptor mellett elhelyezkedő membrán szerkezete, mind a receptor egyes komponensei, ami megváltoztathatja a receptor érzékenységét a rá jellemző biológiailag aktív anyaggal szemben. Azokban az esetekben, amikor a receptor érzékenysége növekszik egy biológiailag aktív anyaggal szemben, beszéljünk érzékenyítés(a lat. sensus - érzés) vagy kb érzékenyítés(a lat. sensibilis - érzékenység), és azokban az esetekben, amikor a receptor érzékenysége csökken, beszéljünk desensitizationreceptor. Az alloszterikus hatás sajátossága abban rejlik, hogy az ilyen típusú hatásmechanizmussal rendelkező gyógyszerek nem közvetlenül érintik az idegimpulzus átadását, hanem módosítják azt a kívánt irányba. Például a szorongásgátlók (szorongásgátló szerek; szinonimák: nyugtatók) kémiai szerkezetükben a benzodiazepin származékainak működési mechanizmusa a posztszinaptikus benzodiazepin receptorok alloszterikus gerjesztésének jelenségén alapul. Ez utóbbi gerjesztése elősegíti a gamma-amino-vajsav gátló posztszinaptikus receptorok (GABA-receptorok) aktiválódását, amely klinikailag a neurotikus betegségek tüneteinek, például szorongás, szorongás, félelem stb. Azokat a receptort, amelyek kölcsönhatásba lépnek, biológiailag aktív anyag vagy gyógyszer bármilyen módon megváltoztatja a célsejt funkcionális állapotát, nevezzük specifikus. A specifikus receptorokon kívül, úgynevezett nem specifikusgyógyszer-receptorok számára. A szakorvosi szakirodalomban ezeket a receptorokat a drogok "veszteségének" helyének is nevezik. Ha ilyen gyógyszerekkel érintkeznek, a gyógyszereknek nincs biológiai hatása, de maguk biológiailag inaktívvá válnak. Az ilyen típusú receptor például a plazmafehérjékben, különösen a vízben oldódó fehérjékben - az albuminban található receptorokként szolgálhat. A receptorok szerkezete meglehetősen bonyolult, de ezek többsége protein makromolekulák vagy glikoproteinek, amelyek tartalmazhatnak ionokat, lipideket, nukleinsavakat stb. Receptor, azaz az azt alkotó fehérje makromolekulát egy specifikus, az egyes receptorokra specifikus, kémiai csoportjainak térbeli elrendezése jellemzi. A receptort alkotó makromolekula integrálható (bemeríthető) a citoplazmatikus membrán lipid kettős rétegébe vagy lokalizálható a sejt belsejében. A sejtreceptor fő funkciója egy endogén biológiailag aktív anyagon és / vagy gyógyszereken keresztül továbbított kémiai jel „felismerése”, és megfelelő biokémiai és / vagy biofizikai sejtválaszvá történő átalakítása. Korábban azt hitték, hogy a gyógyszerek vagy az endogén biológiailag aktív anyagok kölcsönhatásba lépnek a „kulcs és zár” típusú receptorokkal, azaz a receptornak olyan szerkezete van, amely lehetővé teszi a gyógyszer számára, hogy megtalálja az „Ön” receptort, csatlakozzon hozzá, és minthogy „bekapcsolja” és „kapcsolja ki”. Az orvostudomány fejlődésével azonban nyilvánvalóvá vált, hogy ez nem teljesen igaz. Jelenleg az extracelluláris szignálok intracelluláris, sejtfunkciót szabályozó molekuláris átalakításának molekuláris folyamatait már elég jól megvizsgálták. mechanizmusok, amelyek hatással vannak az endogén biológiailag aktív anyagok vagy gyógyszerek receptorokkal való kölcsönhatására. Amikor kölcsönhatásba lép egy endogén biológiailag aktív anyag és / vagy egy hasonló hatóanyag receptoraival konformáció- a fehérje makromolekulájának térbeli változása, amely kiváltó különféle intracelluláris folyamatokban, amelyek meghatározzák a célsejt meditátorra és / vagy gyógyszerre adott válaszát. Például a hörgők simaizmok b2-adrenoreceptorának aktiválása egy b2-adrenostimulátor fenoterol hatására az adenilát-cikláz enzim aktivitásának növekedéséhez vezet, amely hozzájárul a ciklikus adenozin-monofoszfát (cAMP) felhalmozódásához a sejtben és ennek eredményeként a sejtek relaxációjához. Általános biológiai értelemben a celluláris receptorokat szigorúan specializált sejtek „szenzoros szervének” lehet tekinteni, amelyek révén érzékelik például a központi idegrendszerből és / vagy az endokrin rendszerből származó „információt”. A receptorkészülék fontos szerepe ellenére a receptorok a sejtmembrán csak jelentéktelen részét foglalják el. Például egy sejt M-kolinerg receptor berendezése a felületének legfeljebb 1/6 000-ét foglalja el. A gyógyszerek és a receptor közötti kölcsönhatás jellemzőinek tanulmányozása egyrészt lehetővé teszi, hogy megértsük annak molekuláris mechanizmusának alapjait, másrészt információt nyújt arról, hogy milyen változtatásokat kell végrehajtani a gyógyszerek szerkezetében annak fokozása érdekében, hogy kölcsönhatásba lépjenek a receptorral, azaz - lehetővé teszi új, nagyon hatékony gyógyszerek célzott szintézisét. Fiziológiai körülmények között a különböző celluláris receptorok nem működnek egymástól függetlenül, hanem állandó kölcsönhatásban vannak egymással, ezáltal szabályozva a sejt fajlagos aktivitását. Például, ha a szív b-adrenerg receptorokat endogén norepinefrinnel aktiválják, különösen a szív összehúzódások számának növekedése, és a szívsejtek M-kolinerg receptorának aktiválása endogén acetilkolin által, éppen ellenkezőleg, csökkenti a szív összehúzódások számát. A pre- és posztszinaptikus receptorok felfedezése nagyban hozzájárult a gyógyszerek receptor-hatásmechanizmusainak megértéséhez. Balvások(görögül szinopszis - kapcsolat) egy speciális érintkezési zóna az idegsejtek vagy a test más ingerlékeny szerkezete között, amely biztosítja a beérkező információk továbbítását és információs jelentőségének megőrzését. A szinapszis szerkezetének és funkcionális szerepének tanulmányozása a 19. század végén kezdődött. Miután a spanyol histológus S. Ramon-i-Cajal (S. Ramon a Cajalnál) egy speciális átviteli rendszer jelenlétét javasolta a központi idegrendszerben. A szinapszis 1897-ben kapta a nevét, amikor Ch. Sherrington angol fiziológus javasolta, hogy ez a kifejezés az idegsejtek közötti kapcsolatfelületre utaljon. Jelenleg a szinapszisok három típusa létezik: 1) „elektromos” szinapszisok, amelyekben az információ továbbítódik egy preszinaptikus membránról származó elektromos jel átvitelével. Ezt a fajta szinapszist hívják efaps(görögül ephapsis - szoros érintkezés); 2) "kémiai" szinapszis, amelyben az információ speciális biológiailag aktív anyagokon keresztül továbbadódik - neurotranszmitterek(görögül neuron - ideg és lat. közvetítő - mediátor; szinonimák: közvetítő); 3) „vegyes” szinapszis, amelyben az információt mind kémiai, mind elektromos úton továbbítják. A szinapszis működését befolyásoló gyógyszerek túlnyomó többségének farmakológiai hatásai úgy valósulnak meg, hogy a kémiai szinapszisokban a jelátvitel egy adott szakaszára, azaz a második típusú szinapszisban. A kémiai szinapszákat általában az alábbiak szerint osztályozzák az idegimpulzusokat közvetítő neurotranszmitterek: a szinapszisokat, amelyekben az acetilkolin mediátorként működik, kolinergnek nevezzük; szinapszis, amelyben az adrenalin és a norepinefrin mediátorként működik, adrenergnek nevezzük; szinapszisokat, amelyekben az ATP és az adenozin mediátorként hatnak, purinergnek nevezik; olyan szinapszákat, amelyekben a gamma-aminó-vaj mediátorként működik, GABA-ergikusnak nevezik stb. A szinapszis felépítése jelenleg jól ismert. A szinapszis egy idegsejt (axonvég) preszinaptikus folyamatából és egy “jelet” fogadó készülékből áll, amely az effektor („végrehajtó”) sejt membránján helyezkedik el.
Farmakodinámiák - farmakológiai hatások, a hatásmechanizmusok, a hatás lokalizálása, a gyógyszerek hatás típusai.
Farmakológiai hatások gyógyszer anyag - a szervek, a testrendszer aktivitásában bekövetkező változások, amelyeket az anyag okoz (például megnövekedett szív összehúzódások, csökkent vérnyomás, serkenti a mentális tevékenységet, kiküszöbölik a félelmet és a feszültséget, stb.).
Minden anyag számos, a neki jellemző farmakológiai hatást vált ki. Mindegyik esetben csak a hatóanyag bizonyos hatásait alkalmazzák, amelyek meghatározása a következő: a fő hatásokat. A fennmaradó (fel nem használt, nemkívánatos) farmakológiai hatásokat nevezzük kedvezőtlen.
A hatásmechanizmusok gyógyszerek - az anyagok farmakológiai hatásainak módjai nagyon változatosak. A cselekvési mechanizmusok fő lehetőségei a következőkkel kapcsolatos tevékenységeket foglalják magukban:
- specifikus receptorok;
- enzimek;
- ioncsatornák;
- szállítási rendszerek.
A legtöbb gyógyszer befolyásolja specifikus receptorok. Ezeket a receptort leggyakrabban funkcionálisan aktív fehérjemolekulák képviselik, amelyek kölcsönhatása biokémiai reakciókat vált ki, amelyek farmakológiai hatásokat eredményeznek.
Vannak specifikus receptorok, amelyek kapcsolódnak a sejtmembránokhoz (membránokhoz), és intracelluláris receptorok (citoplazmatikus, nukleáris).
A membránreceptorokat (a citoplazmatikus membrán receptorait) fel kell osztani:
- receptorok, amelyek közvetlenül kapcsolódnak ioncsatornákhoz;
- közvetlenül az enzimekkel konjugált receptorok;
- receptorok, amelyek kölcsönhatásba lépnek G-fehérjékkel.
K ioncsatornákkal közvetlenül kapcsolt receptorokEzek közé tartoznak különösen az Ν-kolinerg receptorok és a GABA A-receptorok.
Amikor stimulálják az Ν-kolinerg receptorokat (nikotin-érzékeny kolinerg receptorok), akkor a hozzájuk közvetlenül kapcsolódó nátriumcsatornák kinyílnak. Az Ν-kolinerg receptorok stimulálása Na + csatornák felfedezéséhez, a Na + -ionok belépéséhez a sejthez, a sejtmembrán depolarizációjához és izgalmas hatáshoz vezet.
A GABA A receptorok közvetlenül kapcsolódnak klórcsatornákhoz. A GABA A receptorok stimulálása a Cl - csatornák felfedezéséhez, a Cl - ionok belépéséhez, a sejtmembrán hiperpolarizációjához és a gátló hatáshoz vezet.
K receptorok, amelyek közvetlenül kapcsolódnak az enzimekhezIde tartoznak különösen az inzulinreceptorok, amelyek közvetlenül konjugáltak tirozin-kinázzal.
G-protein receptorok - M-kolinerg receptorok (muszkarin-érzékeny kolinerg receptorok), adrenerg receptorok, dopamin receptorok, opioid receptorok stb.
A G-fehérjék, azaz a GTP-kötő fehérjék a sejtmembránban lokalizálódnak, és a-, β- és γ-alegységekből állnak. Amikor egy gyógyszer kölcsönhatásba lép egy receptorral, a G-protein α-alegysége kötődik a GTP-hez (GTP) és enzimekre vagy ioncsatornákra hat.
Az egyik receptor kölcsönhatásba lép számos G-fehérjével, és a G-protein α-alegységének minden egyes komplexe a GTP-vel több enzimmolekulára vagy több ioncsatornára hat. Így megvalósul az erősítő (erősítő) mechanizmusa: amikor egy receptor aktiválódik, sok enzimmolekula vagy sok ioncsatorna aktivitása megváltozik.
Az egyik első olyan szívfehérje, amely a szív β 1 -adrenoreceptoraival kapcsolatos. Amikor aktiválódik a szív szimpatikus beidegzése, a β 1 \u200b\u200b-adrenoreceptorok izgatottak; az adenilát-cikláz G-fehérjék útján aktiválódik; ATP-ből cAMP képződik, protein-kinázt aktiválnak, amelynek hatására a Ca2 + -csatornákat foszforilálják és megnyitják.
A szinoatriális csomópont sejtjeibe jutó Ca 2+ -ionok növekedése felgyorsítja az akciópotenciál negyedik fázisát, a generált impulzusok frekvenciája növekszik - a szív összehúzódások egyre gyakoribbak.
A Ca 2+ csatornák felfedezése a működő szívizom rostjaiban növeli a Ca 2+ koncentrációját a citoplazmában (a Ca 2+ bevitel elősegíti a Ca 2+ felszabadulását a szarkoplazmatikus retikulumból). Ca 2+ -ionok kapcsolódnak a troponin C-hez (a troponin-tropomyosin egyik alkotóeleme); Így a troponin-tropomyosin gátló hatása az aktin és a miozin kölcsönhatására csökken - a szív összehúzódása fokozódik (10. ábra).
Ábra. 10. A szív összehúzódások fokozásának és erősítésének mechanizmusa a β 1 \u200b\u200b-adrenoreceptorok stimulálása során. AC - adenilát-cikláz; PC - protein-kináz; CA - kínai csomópont; TTM - troponin-tropomyosin.
Amikor a szív (vagus idegek) parasimpátikus beidegződése aktiválódik, az M 2 -kolinerg receptorok gerjesztésre kerülnek, és a G-fehérjék révén gátolják az adenilát-cikláz - a szív összehúzódások gyengülnek és gyengülnek (elsősorban a pitvari összehúzódások gyengülnek, mivel a kamrák parasimpátikus beidegzése viszonylag gyenge).
Így a G-fehérjék stimuláló és gátló hatást gyakorolhatnak az adenilát-ciklázra. A stimuláló G-fehérjéket Gs-fehérjéknek (stimulálni) és gátló G-fehérjéknek (gátlásoknak) neveztük (11. ábra).
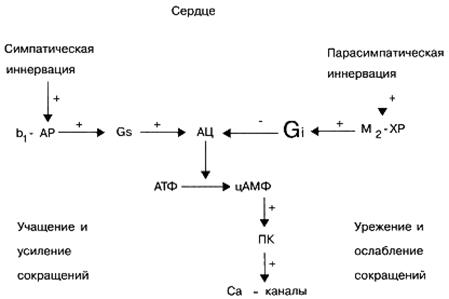
Ábra. 11. A szívösszehúzódások gyakoriságának és erősségének változásának mechanizmusa a szimpatikus és a parasimpatikus beidegzés stimulálása során.
A kolera toxin aktiválja a C fehérjéket (ez az adenilát-cikláz aktiválásához vezet, és a koleraban a folyadék kiválasztása a bél hámán keresztül történik).
A pertussis toxin aktiválja a G i fehérjéket.
Amikor az M1-kolinerg receptorok, az M3-kolinerg receptorok, az α1 -adrenoreceptorok gerjesztik a Gq fehérjéket, aktiválódik a foszfolipáz C, amely hozzájárul az inozitol-1,4,5-trifoszfát és a diacil-glicerin képződéséhez foszfatidil-inozitol-4,5-difoszfátból .
Az inozitol-1,4,5-trifoszfát hatással van a rá érzékeny sarkoplazmatikus retikulum membrán receptoraira és serkenti a Ca 2+ ionok felszabadulását a sarkoplazmatikus retikulumból (12. ábra). Az erek αι-adrenoreceptorok stimulációjával ez az erek simaizmok összehúzódásához és az erek szűkítéséhez vezet (13. ábra).

Ábra. 12. A foszfolipáz C hatása a citoplazmatikus C a2 + szintjére.
A receptorok agonistákkal szembeni érzékenysége és a receptorok száma folyamatosan változik. Tehát, miután a β 1 \u200b\u200b-adrenoreceptorokat stimulálta egy β 1 -adrenoreceptor agonistával, egy speciális receptor kináz által foszforilálódik, kötődik a β-arrestin fehérjéhez, és ebben a komplexben elveszíti képességét, hogy kölcsönhatásba lépjen a G-fehérjékkel (receptor deszenzibilizáció). A β 1 -adrenoreceptorok és a β-arrestin komplexét a sejt endocitózissal (receptorok internalizálása) felszívja, és az endoszómák és a lizoszómák fogják el. Az endoszómákban a β1-resztin molekulák leválódnak a receptoroktól, amelyek újra integrálódnak a sejtmembránba; a receptor agonistákkal szembeni érzékenysége helyreáll (receptor resenzitizáció). A lizoszómákban a receptor molekulák elpusztulnak (lefelé történő szabályozás) (14. ábra).

Ábra. 13. Az erek simaizmok összehúzódásának mechanizmusa a szimpatikus beidegzés stimulálása során. FLS - foszfolipáz C; FIF 2 - foszfatidil-inozitol-4,5-difoszfát; Ha 3 - inozitol-1,4,5-trifoszfát; CP - szarkoplazmás retikulum; MLCK - a miozin könnyű láncának kinázai.

Ábra. 14. A β-adrenerg receptorok szenzibilizációja és alulszabályozása.
K intracelluláris receptorok a kortikoszteroid és a nemi hormon receptorok is ide tartoznak. Különösen a glükokortikoid receptorok lokalizálódnak a sejtek citoplazmájában. Miután a glükokortikoidot citoplazmatikus receptorokkal kombinálják, a glükokortikoid - receptor komplex behatol a magba és befolyásolja a különféle gének expresszióját.
Az anyagoknak a receptorokhoz való kötődésének képességét (az anyagok hajlandóságát receptorokhoz történő kötődésre) a " affinitás”. Ugyanazon receptorokhoz viszonyítva a különböző anyagok affinitása eltérő lehet. Az affinitás jellemzéséhez használja a mutatót pKD a disszociációs állandó negatív logaritmusa, azaz az anyag koncentrációja, amelyen a receptorok 50% -a el van foglalva.
Belső tevékenység - az anyagok képessége a receptorok stimulálására; meghatározta farmakológiai hatásreceptor aktiválással jár.
Normál körülmények között nincs közvetlen kapcsolat az affinitás és a belső aktivitás között. Egy anyag elfoglalhatja az összes receptort, és gyenge hatást okozhat, és fordítva: egy anyag elfoglalhatja a receptorok 10% -át, és ennek a rendszernek a maximális hatását okozhatja.
agonisták - affinitású és belső aktivitású anyagok.
Teljes agonisták affinitással és maximális belső aktivitással rendelkeznek (egy adott rendszer maximális hatását okozhatják), még akkor is, ha a specifikus receptorok részét képezik.
Részleges (részleges) agonisták affinitással rendelkeznek és a maximális belső aktivitásnál kevesebbek (csak a maximális hatásokat okozhatják, még akkor is, ha a specifikus receptorok 100% -át elfoglalják).
antagonistákaffinitással rendelkeznek, de nem rendelkeznek belső aktivitással, és nem zavarják a teljes vagy részleges agonisták működését (kiszorítják az agonistákat a receptorokkal való kommunikációtól).
Ha az antagonista hatását az agonista dózisának növelésével kiküszöböljük, akkor ezt az antagonistát versenyképesnek nevezzük.
A részleges agonisták lehetnek a teljes agonisták antagonistái. Teljes agonista hiányában a részleges agonista stimulálja a receptorokat és gyenge hatást vált ki. Ha egy komplett agonistával kölcsönhatásba lép, a parciális agonista receptorokat foglal el és gátolja a teljes agonista hatását. Ebben az esetben a teljes agonista hatása gyengült.
Például a pindolol, egy részleges β-adrenoreceptor-agonista, gyenge tachikardiat okoz, a szimpatikus beidegződésnek a szívre gyakorolt \u200b\u200bhatásának hiányában. De a szimpatikus beidegződés fokozódásával a pindolol valódi β-blokkolóként működik és bradycardia kialakulását okozza. Ennek oka az a tény, hogy a parciális agonista pindolol gyengíti a noradrenalin mediátor hatását, amely teljes agonista a szív β-adrenerg receptoraival szemben.
Antagonista agonisták - olyan anyagok, amelyek ugyanazon receptorok altípusain eltérően hatnak: stimulálnak egyes receptor altípusokat, és blokkolnak másokat. Például a kábítószer-fájdalomcsillapító nalbufinnak különböző hatása van az opioid receptor altípusokra. A nalbufin stimulálja a κ receptorokat (és ennélfogva csökkenti a fájdalomérzékenységet), és a μ receptorok blokkolják (és ezért a gyógyszerfüggőség szempontjából kevésbé veszélyesek).
Példa az anyagok hatására enzimek előfordulhat, hogy az antikolineszteráz szerek blokkolják az acetilkolinészterázt (egy enzim, amely lebontja az acetilkolint), és ezáltal fokozza és meghosszabbítja az acetilkolin hatását.
Ismert gyógyhatású anyagok, amelyek stimulálják vagy blokkolják ioncsatornák sejtmembránok, vagyis olyan csatornák, amelyek szelektíven vezetnek Na +, K +, Ca 2+ ionokat (nátrium, kálium, kalcium csatornák) stb. Például:
A helyi érzéstelenítők blokkolják a Na + csatornákat;
I. osztályú antiaritmiás szerek (kinidin, lidokain) blokkolják a Na + csatornákat;
A minoxidil aktiválja a K + csatornákat;
A szulfonilkarbamid-származékok csoportjából származó hipoglikémiás szerek blokkolják az ATP-függő K + csatornákat;
Verapamil, a nifedipin blokkolja a Ca 2+ csatornákat.
Példa az anyagok hatására közlekedési rendszerek lehet egy akció:
Rezerpin (blokkolja a dopamin és norepinefrin veszikuláris felvételét);
Szívglikozidok (gátolják a Na + / K + -ATPáz-t);
Triciklusos antidepresszánsok (blokkolja a norepinefrin és a szerotonin fordított idegrendszeri felvételét);
Protonpumpa-blokkolók (omeprazol, stb.).
Más hatásmechanizmusok is lehetségesek. Például, a vizelethajtó mannit növeli a vizeletürítést azáltal, hogy növeli az ozmotikus nyomást a vese tubulusokban. Az antiateroszklerotikus szer - a kolestipol - megköti (szekvesztrálja) az epesavakat, megakadályozza azok felszívódását a bélben, ezért aktiválódik az epesavak képződése a koleszterinből a májban, és csökken a májsejtek koleszterinszintje.
A különféle gyógyszerek hatásmechanizmusait különféle mértékben vizsgálták. Tanulmányozásuk során a cselekvési mechanizmusokkal kapcsolatos ötletek nemcsak bonyolultabbá válhatnak, hanem jelentősen megváltozhatnak is.
A " akció lokalizációja"Egyes gyógyászati \u200b\u200banyagok elsődleges hatása (i). Például a szívglikozidok főként a szívre hatnak.
A " cselekvési típusok»Kapcsolódás a helyi és általános (rezorpciós) tevékenységekhez, reflex akció, elsődleges és másodlagos fellépés, közvetlen és közvetett cselekvés.
A helyi fellépés példája lehet a helyi érzéstelenítés.
A legtöbb gyógyszernek általános (rezorpciós) hatása van, amely általában egy anyag vérben történő felszívódása (felszívódása) és a szervezetben történő eloszlása \u200b\u200bután alakul ki.
Mind helyi, mind rezorpciós hatásokkal az anyagok különféle érzékeny receptorokat gerjeszthetnek és reflex reakciókat válthatnak ki.
A gyógyászati \u200b\u200banyag fő hatása a hatása, amelyet minden esetben alkalmaznak. Az összes többi hatást a mellékhatások megnyilvánulásaiként értékelik.
A gyógyászati \u200b\u200bhatóanyagok bizonyos szerveket kifejthetnek közvetlen fellépés. Ezenkívül a drogok hatása közvetett is lehet. Például a szívglikozidok közvetlen hatással vannak a szívre, de javítva a szív működését, ezek az anyagok növelik a vérellátást és más szervek működését (közvetett hatás).