Pharmacodynamics
Pharmacodynamics is a section of general pharmacology that studies the features of the action of drugs on the body. Namely, pharmacodynamics studies:
mechanisms of action of drugs;
final pharmacological effects;
dependence of the action of drugs on various conditions;
the effects of drugs upon repeated administration;
combined action of drugs;
drug incompatibility;
side effects medicinal substances.
The mechanisms of action of drugs are the ways in which substances cause pharmacological effects. The main mechanisms of action of drugs include:
Physical.
The mechanism of direct chemical interaction.
Membrane (physico-chemical).
Enzymatic (biochemical).
Receptor.
Direct chemical interaction. This is a rather rare mechanism of action of drugs, the essence of which is that drugs directly interact with molecules or ions in the body. Such a mechanism of action is possessed, for example, by the unitiol drug belonging to the group of antidotes. In case of poisoning with thiol poisons, including salts of heavy metals, unitiol enters into a direct chemical reaction with them, resulting in the formation of non-toxic complexes that are excreted in the urine. Thus, antacids also act, which enter into a direct chemical interaction with hydrochloric acid, lowering the acidity of gastric juice.
Membrane (physicochemical) mechanism. It is associated with the effect of drugs on ion currents (Na +, K +, Cl - and others), which determine the transmembrane electric potential. According to this mechanism, anesthetics, antiarrhythmic drugs, local anesthetics, etc.
Enzymatic (biochemical) mechanism. This mechanism is determined by the ability of some drugs to exert an activating or inhibitory effect on enzymes. The arsenal of drugs with such a mechanism of action is very wide. For example, anticholinesterase drugs, monoamine oxidase inhibitors, proton pump blockers, etc.
Receptor mechanism. In the human body there are highly specific biologically active substances (mediators) that interact with receptors and change the functions of various organs or tissues of the body.
Receptors are macromolecular structures with selective sensitivity to certain chemical compounds. With the interaction of drugs with receptors, biochemical and physiological changes in the body occur, accompanied by one or another clinical effect.
Mediators and drugs that activate receptors and cause a biological effect are called agonists. Medicinal substances that bind to receptors, but do not cause their activation and biological effect, reduce or eliminate the effects of agonists, are called antagonists. Allocate also antagonist agonists - substances that act differently on subtypes of the same receptors: they stimulate some receptor subtypes, and block others. For example, the narcotic analgesic nalbuphine stimulates opioid kappa receptors (therefore, reduces pain sensitivity) and blocks opioid mu receptors (therefore, is less dangerous in terms of drug dependence).
The ability of substances to bind to receptors is referred to by the term “affinity”. In relation to the same receptors, the affinity of different substances can be different.
The following types of receptors are distinguished:
Plasma membrane receptors:
channel type: N-type cholinergic receptors, muscle-type H-cholinergic receptors, GABA receptors;
g-protein receptors: α- and β-adrenergic receptors, M 3 -cholinoreceptors;
integrative type receptors: NO receptor.
Cytosolic.
Mitochondrial.
Nuclear
Channel Type Receptors
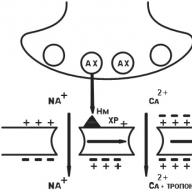
N n nerve type cholinergic receptor (CNS, autonomic ganglia, sinocarotid zone, chromaffin adrenal gland tissue). After the binding of acetylcholine (AX) with H n -cholinergic receptors, Na + channels open and Na rushes into the cell, bearing a positive charge. The postsynaptic membrane is depolarized. There is an action potential that shifts along the membrane of the neuron, opening up electrically dependent Na + channels. A nerve impulse arises in the postganglionic fiber (Fig. 6).
Fig. 6. N n -cholinergic receptor
N m muscle type cholinergic receptor (skeletal muscle cell membranes). The initial processes are similar, but electrically dependent Ca ++ channels open. Ca ++ ions enter the muscle fiber, Ca ++ is released from the sarcoplasmic reticulum. The level of Ca ++ rises, which induces muscle contraction (Fig. 7).

Fig. 7. N m-cholinergic receptor
GABA receptors. These are receptors for γ-aminobutyric acid (GABA). GABA interacts with GABA receptors, in the structure of which there are chloride channels. As a result of receptor stimulation, channels open and chlorine ions (Cl -) freely enter the cell. An increase in the concentration of chlorine ions inside the cell leads to hyperpolarization of the membrane and a decrease in the activity of neurons. It is more difficult to excite such a cell (Fig. 8).
 Fig. 8. GABA receptor:
Fig. 8. GABA receptor:
GABA-R - GABA-receptor, BD-R - benzodiazepine receptor, BR - barbiturate receptor
Receptors associated with G protein
G-proteins, i.e., GTP-binding (guanosine triphosphate-binding) proteins, are localized in the cell membrane and consist of α-, β- and γ-subunits. They (G-proteins) regulate the activity of specific effectors (instant messengers, secondary intermediaries). These messengers can be enzma (adenylate cyclase, phospholipase); channels for potassium, calcium, sodium; some transport proteins. Each cell can have many G-proteins, each of them regulates the activity of various messengers, while changing the function of the cell.
M 3 cholinergic receptor (smooth muscle membranes (MMCs) and exocrine gland cells). Acetylcholine stimulates M 3 -XR bound to the G protein. Phospholipase-C (FLS) is activated, which catalyzes the cleavage of PIDP (phosphatidylinositol diphosphate) into ITP (inositol triphosphate) and DAG (diacylglycerol). ITF entering the cytoplasm of MMC releases Ca ++ from cavelles .

Fig. 9. M 3 -cholinergic receptor
Ca ++ binds to calmodulin, activates myosine kinase (MK), which catalyzes the phosphorylation of myosin light chains, which leads to cell contraction (Fig. 9). Similarly, an impulse is transmitted in the synapses of the secretory glands.
Norepinephrine stimulates α 1 adrenoreceptorBy starting the following chain of events:
Norepinephrine (HA) → α 1 -adrenoreceptor → activation of the α-subunit of G s-protein → activation of PLL → FIDF cleavage → increase in ITF concentration → increase in Ca 2+ concentration in the cell → Ca 2+ binds to calmodulin → myosine kinase is activated → light chains phosphorylate → light chains myosin → myosin interacts with actin → a reduction in MMC develops (Fig. 10).

Fig. 10. α 1 -adrenoreceptor
1 -receptor(fig. 11). Norepinephrine → activates 1 -AP → activation of the α-subunit of G-protein → activation of AC → increase in the formation of cAMP from ATP → increase in the concentration of cAMP in cardiomyocytes → activation of protein kinases → phosphorylation of calcium channel proteins → increase in Ca 2+ entry through channels and increase in Ca concentration 2+ in the cell → an increase in the force of contractions of the heart.

Fig. eleven. 1 receptor
2 -receptor(fig. 12). ON → 2 -AR → activation of the α-subunit of the G-protein → activation of ACs → increased formation of cAMP → stimulated protein kinase → the kinase that catalyzes the phosphorylation of myosine kinase is cleaved, while the latter’s activity is lost → myosin phosphorylation does not occur → MMC relaxation.
The release of HA from nerve endings is regulated by the mediator himself upon excitation of the α 2 -AP presynaptic membrane. The emission of HA decreases.

Fig. 12. 2 receptor
Integrative type receptors
These are receptors that are proteins that penetrate the membrane. In this case, the outer part of the protein plays a receptor role, while the inner part plays a catalytic role (Fig. 13).

Fig. thirteen. Integrative type receptor
Cytosolic receptors
Under physiological conditions, such receptors serve to bind steroid hormones (sex hormones, glucocorticoids). These substances enter the cell and bind to cytosolic receptors there. This complex penetrates the nucleus and changes the work of the genome there. As a result, protein synthesis in the cell changes (Fig. 14).

Fig. 14. Cytosolic receptor
Mitochondrial receptors
In the mitochondria there are also receptors with which medicinal substances interact, such as triiodothyronine hydrochloride, which are analogues of the natural hormone T 3. As a result of this interaction, ATP synthesis increases.
Nuclear receptors
T 3 penetrates the nucleus and there interacts with receptors of this type. As a result, the work of the genome changes and new proteins are synthesized.
Final pharmacological effects (according to Vershinin)
Despite the abundance of drugs, the changes caused by them in the body are of the same type (Fig. 15). The effect of any drug on the organs can be reduced to five main pharmacological effects (according to N.V. Vershinin):
Soothing - reduction to normal of increased organ function (use of sedatives).
Oppression - decrease below the norm of the function of the body (use of drugs for anesthesia)
Paralysis - cessation of reduced organ function (respiratory depression in case of an overdose of narcotic analgesics).
Toning up - strengthening of reduced function to normal (use of β 1 -adrenomimetics).
Excitation - Increasing the function of the organ in excess of the norm (the use of diuretics in case of poisoning, expectorant drugs).

Fig. 15. Final pharmacological effects
Types of action of drugs
The main thing is secondary.
Reversible, irreversible.
In few cases, the therapeutic goal requires irreversible turning off a structure from its function. This applies, for example, to most antimicrobial, antitumor agents that are able to form strong (covalent) bonds with the elements of DNA helix cells ("crosslinking") or bacterial enzymes, as a result of which the cells lose their ability to reproduce.
Direct, indirect (indirect).
A special case of an indirect action is reflex act. For example, vasodilation and improvement of trophic tissue as a result of irritation of the ends of the sensory nerves of the skin.
Selective, non-selective.
Local, resorptive.
Resorptive (systemic) action develops after absorption of the drug into the blood. The vast majority of drugs have this effect.
Factors Affecting Pharmacokinetics
and pharmacodynamics
I. External factors
Environment:
season (in the summer after taking tetracycline, sunburn is possible (the drug increases the sensitivity of the skin to ultraviolet light));
ambient temperature (in hot weather, a stronger effect of drugs that depress the central nervous system is manifested);
partial pressure of O 2 (caused by epinephrine (adrenaline) tachycardia is better tolerated at high partial pressure of O 2).
Properties of drugs:
solubility (soluble Ba 2 CO 3 is toxic, and insoluble Ba 2 SO 4 is not toxic);
radicals (substitution of the CH 3 - group at the nitrogen atom in the morphine molecule with the -CH 2 -CH \u003d CH 2 - group (naloxone) leads to the appearance of properties antagonistic to morphine in the substance);
isomerism (levorotatory isomer of propranolol (anaprilin) \u200b\u200bis 40-60 times more potent than dextrorotatory);
polarity (polar molecules are usually poorly soluble in lipid membranes, therefore, poorly absorbed and poorly penetrate through cell membranes).
The intake of drugs in the body:
dosage form (medicine in liquid form has greater bioavailability, the effect begins faster, and it is more pronounced);
route of administration (with intravenous administration The drug acts faster and stronger than when taken by mouth, the duration of its action is shorter);
dose (with increasing dose (up to a certain limit), the strength of the action of drugs increases);
a combination of drugs (possibly weakening, summing up, enhancing the effects of combination drugs, and sometimes strengthening some and weakening other effects of drugs);
duration of administration (with prolonged use barbiturates their effect is reduced, as their metabolism in the liver is accelerated).
II. Intrinsic factors
Biological object:
species features (rabbits easily tolerate lethal doses of atropine for humans);
ethnic features (in people of the Mongoloid race, alcohol dehydrogenase deficiency is more common and, as a result, their sensitivity to ethanol is higher than that of Europeans);
age (in newborns and young children, the ability of the liver to metabolize drugs is low, the kidneys do not fully function and the fluid content in the body is higher than in adults; in elderly people, the metabolism of drugs is reduced, and kidney function decreases with age);
gender (the elimination of many drugs in men is faster than in women, since it is believed that male sex hormones activate liver enzymes);
genotype (in people with defective (inactive) pseudocholinesterase, respiratory arrest after administration of the muscle relaxant suxamethonium (dithilin) \u200b\u200bdoes not last 2-3 minutes, as in most patients, but 2-3 hours or more due to a sharp decrease in the rate of destruction of suxamethonium (idiosyncrasy) );
phenotype (in obese people, lipophilic drugs (phenobarbital, etc.) are cumulated to a greater extent than in thin ones).
The physiology of the body:
nutrition (food can have a significant effect on the pharmacokinetics of the drug, most often slow down and reduce the absorption of drugs);
pregnancy (many drugs that cross the placental barrier can affect fetal development);
lactation (antibiotics used by the mother and the child receive milk, which can, for example, cause him to have dysbiosis);
stress (excited people are most sensitive to substances of a stimulating effect);
circadian rhythms (sulfonamides are more slowly excreted by the kidneys at night when urine pH decreases).
Pathological conditions:
diseases (in patients with cirrhosis of the liver, drugs such as barbiturates and chlorpromazine can cause an unusually long effect);
alcoholism (ethyl alcohol enhances the effect on the central nervous system anxiolytics, anticonvulsants and antidepressants);
smoking (biotransformation of many drugs in the body of smokers is faster (reduced effectiveness of combined oral contraceptives in women smokers)).
The dose is the amount of drugs introduced into the body and causes any pharmacological effect.
Usually, doses are expressed in units of mass, volume (grams, milliliters). In units of action (ED), certain drugs of biological origin with intermittent activity (hormones, antibiotics, heparin, etc.) are dosed.
Dose classification
By time of administration:
one-time;
daily allowance;
course.
By force of action:
medical or therapeutic (minimal, medium, higher);
toxic (minimal, medium, lethal).
A saturating dose is the dose by which it is possible to create the necessary concentrations of drugs in the tissues (for example, in the treatment of cardiac glycosides).
Maintenance dose - the dose with which you can maintain the plasma and tissue concentration of drugs, making up for the loss of the drug during the elimination process (for example, digitalization).
A shock dose is a dose that allows you to create the optimal concentration of the drug necessary for its competition with a certain endogenous substrate (for example, the shock dose of sulfanilamides necessary for competition with para-aminobenzoic acid (PABA) for a place in the structure of a folic acid molecule at the stage of its synthesis). , drugs are prescribed in medium therapeutic doses, which in most patients have the optimal therapeutic effect without toxic effects. Typically, such a dose is 1/2 or 1/3 of the maximum therapeutic.
The dose range between minimal therapeutic and minimal toxic is called breadth of therapeutic effect(SHTD) . The greater the breadth of therapeutic action, the safer drugs (Fig. 16).
SHTD

Fig. 16. The breadth of therapeutic action
1 - minimum therapeutic dose, 2 - average therapeutic dose,
3 - maximum therapeutic dose, 4 - minimum toxic dose
Therapeutic index (TI) is the ratio of the toxic dose to the average therapeutic dose, determined by the formula:
where TD 50 is the dose causing poisoning in 50% of patients;
ED 50 - the average therapeutic dose, i.e. the dose that causes a therapeutic effect in 50% of patients.
The more TI, the safer the drugs. In order for the drug to be safe, its TI should be more than 3.
In children and seniors, the dosage of drugs has its own characteristics, which is associated with the physiological differences of these groups.
Features of the child's body:
failure of the metabolizing function of the liver (therefore, drugs are more toxic);
the skin and mucous membranes are abundantly vascularized (therefore drugs are absorbed better than in adults);
The BBB is more permeable (this creates a relatively large concentration of drugs in the brain);
high water content in tissues;
less adipose tissue
To a lesser extent, drugs bind to plasma proteins (this can lead to toxic reactions, since the free (active) fraction increases);
excretory function of the kidneys is reduced (this leads to a longer action of drugs).
In view of the presence of sufficiently significant differences in the pharmacodynamics and pharmacokinetics of drugs in children and adults, a simple dose-proportional reduction in the dose of adults proportional to age is unacceptable when calculating the dose of a drug for a child, because can lead to unpredictable consequences.
The dose in children is calculated per 1 kilogram of body weight, per year of life, per body surface area. For instance:
Dose of the child \u003d Adult dose × weight of the child / 70 kg
Physiological characteristics of senile people:
violation of the metabolism of drugs in the liver as a result of atrophic and dystrophic changes;
low water content in the body and a higher content of adipose tissue;
a decrease in plasma proteins (this leads to an increase in the free fraction of drugs);
progressive decrease in renal excretory function;
The central nervous system and the cardiovascular system are more sensitive to the action of drugs.
Side (undesirable) effects of drugs
Undesirable effects mean any reactions to drugs that are harmful to the body that occur when they are used to treat, diagnose or prevent diseases. Adverse reactions occur from 1 to 30% of cases of the use of drugs in clinical practice. There are drugs, the use of which very often causes undesirable reactions. These include antibiotics, glucocorticoids, non-steroidal anti-inflammatory drugs, antiepileptic, antitumor and other drugs. Undesirable effects of drugs can be divided into several groups.
Undesirable effects associated with the therapeutic concentration of drugs in the blood:
allergic reactions;
pseudo-allergic reactions;
genetically determined reactions (idiosyncrasy);
syndrome of mental and physical dependence.
Pseudo-allergic reactions (anaphylactoid) are characterized by the direct effect of the drug on the mast cell, without IgE synthesis. Unlike allergic reactions, they are dose dependent. The patient, as a rule, does not have a burdened allergic history. Pseudo-allergic reactions can be caused by ampicillin, iodine-containing radiopaque substances, local anesthetics, etc.
Idiosyncrasy - genetically determined intolerance to drugs. Genetic reactions cannot be predicted. They are associated with hereditary defects of enzyme systems or with hereditary metabolic diseases.
For example, pseudocholinesterase deficiency is accompanied by a suppression of the destruction of dithilin (which leads to prolonged muscle relaxation). The deficiency of glucose-6-phosphate dehydrogenase is accompanied by a decrease in the activity of a number of reducing enzymes (glutathione reductase, etc.). The ingress of oxidizing drugs (sulfonamides, nitrofurans, antimalarial drugs - quinine, chingamine, primaquine, etc.) into the body leads to the formation of hemolysis of red blood cells and the formation of methemoglobin. The hereditary abnormality of the sarcoplasmic reticulum is accompanied by impaired calcium fixation on actomyosin and the general acid-base state when halotane, barbiturates and other drugs are used, which are mainly used in anesthetic practice. Malignant hyperthermia occurs, which can lead to the death of the patient.
Mental and physical dependence (addiction). Drugs are caused by drugs such as opium and its alkaloids (morphine, codeine, heroin), promedol, cocaine, amphetamine, ethanol, some barbiturates, etc.
Euphoria is the root cause of uncontrolled drug use or development mental addiction. Euphoria is characterized by the disappearance or dullness of unpleasant emotions, feelings of fear, anxiety. The desire to experience euphoria again is the cause of psychic dependence.
Physical addiction associated with the appearance of withdrawal syndrome (withdrawal or deprivation syndrome): chills, hyperthermia, sharp fluctuations in blood pressure, muscle and joint pain, vomiting, anxiety, hostility, insomnia. Moreover, the number and intensity of symptoms is associated with the degree of physical dependence.
Perhaps the mechanism for the development of physical dependence is due to the fact that narcotic analgesics, activating opiate receptors by the feedback principle, inhibit the release of endogenous opiate peptides, gradually replacing their activity. As a result of the abolition of analgesics, there is a failure of the previously introduced analgesic and endogenous peptide. Cancellation syndrome develops.
Undesirable effects associated with the toxic concentration of drugs in the blood (mainly characteristic of drugs with a narrow breadth of therapeutic effect):
nephrotoxicity (aminoglycosides);
ototoxicity (prolonged use of aminoglycosides can lead to hearing loss, up to the development of irreversible deafness);
hematotoxicity (the antibiotic levomycetin has a depressing effect on the hematopoietic system);
neurotoxicity (an antimicrobial drug from the group of fluoroquinolones lomefloxacin causes insomnia, headache);
gastrotoxicity (salicylates with prolonged use can lead to peptic ulcer disease);
hepatotoxicity (antibiotic-lincosamides cause jaundice with an increase in plasma levels of hepatic transaminases, indicating damage to the liver tissue);
cardiotoxicity (antitumor antibiotics).
Undesirable effects that do not depend on the concentration of drugs in the blood:
dysbiosis;
superinfection;
hypovitaminosis;
immunodeficiency.
Superinfection develops with the use of highly active antibiotics and other antimicrobial agents. Its occurrence is due to the fact that antibiotics suppress microflora that are sensitive to them, and microflora resistant to antibiotics (apathogenic or conditionally pathogenic) begins to multiply intensively and under certain conditions can cause a new disease - superinfection.
Negative effect of drugs on the embryo and fetus
Of particular relevance in modern conditions is the problem of the effect of drugs in both therapeutic and toxic concentrations on the human fetus. Prescribing drugs to pregnant women requires great care, since drugs can penetrate the placental barrier, appear in the blood of the fetus and have negative effects on it.
Such influences include:
Embryotoxic effect.
Teratogenic effect.
Fetotoxic effect.
Teratogenic effect occurs mainly as a result of taking drugs from the 3rd to the 10th week of pregnancy (I trimester). During this period, histo-and organogenesis occurs. The teratogenic effect is a violation of the differentiation of fetal tissues, because of which a child with malformations of the extremities, head, and internal organs can be born. Depending on the characteristics of the defect, the child may be unviable and die soon after birth, or may remain disabled for life.
An example of teratogenic effects is the underdevelopment of the extremities (focomelia) due to the use of thalidomide. The use of androgens during pregnancy leads to masculinization of the female fetus. The use of large doses of tetracyclines is accompanied by the accumulation of the drug in the bones of the fetus and the violation of their development.
Fetotoxic effect - This is the result of a reaction of a ripening or already mature fetus to drugs, which can cause a change in vital functions. For example, indomethacin and some other NSAIDs cause closure or narrowing of the ductus arteriosus. Aminoglycoside antibiotics cause ototoxicity. Anticoagulants can cause bleeding in a newborn. The use of antithyroid drugs is accompanied by the development of goiter. These toxic reactions can cause severe pathology of the fetus and newborn and increase perinatal mortality in children.
The phenomena developing at repeated introduction of drugs
In clinical conditions, there are not many cases when drugs are used once. This is encountered in rendering emergency care. Most often, drugs are prescribed again. In this case, the following types of reactions can be observed.
Cumulation is the accumulation of a substance in the body ( material cumulation) or its effects ( functional cumulation) Probability material cumulation is the higher, the slower the drug is inactivated in the body and the stronger it binds to the biosubstrate in the tissues. Cumulation is always dangerous due to the rapid increase in the number and severity of various complications and toxic reactions. Barbiturates, cardiac glycosides, etc. are most prone to cumulation. With functional cumulation, the increase in the therapeutic effect, which becomes intoxication, overtakes the physical accumulation of the drug in time (it may not be). So, with alcoholism, increasing changes in the function of the central nervous system can lead to the development of delirium tremens. In this case, the substance (ethyl alcohol) is rapidly oxidized and does not linger in the tissues. Only its neurotropic effects are summarized.
Tolerance (addictive) - this is a gradual weakening (until complete loss) therapeutic action drug with prolonged use. Tolerance can have different causes and usually develops in parallel with all representatives of this pharmacological group. It can be a consequence of the following reactions:
increase or decrease in the number of receptors;
enhancing the functioning of homeostatic regulatory mechanisms that compensate for the shift caused by the drug (for example, an increase in blood pressure-lowering blood vessel vasodilator in hypertensive patients as a result of fluid retention, increased heart rate, the inclusion of other mechanisms for increasing vascular tone);
accelerated inactivation of the drug as a result of the induction by him or another chemical factor of microsomal enzymes.
Attempts to overcome addiction by simply increasing the dosage of the same drug are ineffective and fraught with the development of complications of drug therapy.
Tachyphylaxis - an option of quick tolerance, when addiction occurs quickly, within a few hours or days. For example, tolerance to ephedrine develops already on the second administration of the drug.
Withdrawal syndrome occurs when the drug is suddenly stopped in the following cases:
after the termination of the usual pathogenetic pharmacotherapy (for example, exacerbation of IHD - nitrates, β-blockers);
with the abolition of drugs that can cause withdrawal symptoms (narcotic analgesics, tranquilizers, psychostimulants);
upon completion of therapy, drugs whose analogues are produced in the body (glucocorticoids, thyroid hormones); taking these drugs can inhibit the production of endogenous hormones, which is accompanied by drug dependence.
Ricochet syndrome (recoil phenomenon) - is a kind of withdrawal syndrome. The essence of the phenomenon is the disinhibition of the regulatory process or a separate reaction, previously suppressed by a drug substance. As a result, there is a kind of supercompensation of the process with a sharp exacerbation of the disease compared with even the pre-treatment level.
The best way to prevent is also the gradual withdrawal of the drug.
Drug addiction (see topic “ Side effects medicinal substances ").
Drug Interactions
Simultaneous administration of several medicines used when there are several problems at the same time. However, in the treatment of one disease, several drugs are often prescribed to increase the therapeutic effect and (or) to reduce side effects.
For example, to increase the analgesic effect of fentanyl, it is combined with droperidol.
To reduce the hypokalemia caused by hydrochlorothiazide, panangin is prescribed (contains potassium asparaginate).
To increase the therapeutic effect and reduce side effects, levodopa is combined with carbidopa.
A type of therapy, when a large number of medications are scientifically unreasonably used to treat a disease, is called polypharmacy.
If one drug enters the body, then about its fate (pharmacokinetics) and its effects (pharmacodynamics) can be concluded in about 90-95% of cases; if two drugs - only in 50% of cases, and if more than three drugs enter the body - up to 10%. To this it should be added that the risk of adverse reactions with the combined use of drugs also increases: by 5% if used up to 5 drugs, by 20% - up to 8 drugs and by 40% - up to 15 drugs.
The interaction of drugs can be undesirable, adverse. Possible weakening of the therapeutic properties of drugs, increasing their side effects or the occurrence of toxic effects. In such cases, they talk about drug incompatibilities.
For example, penicillins have a bactericidal effect on growing microorganisms, and tetracyclines disrupt protein synthesis and inhibit bacterial growth. In this regard, tetracyclines weaken the effect of penicillins.
Drug Interactions may be:
Pharmaceutical
Pharmacological.
For example, it is impossible to combine solutions of aminophylline with solutions of pipolfen or ascorbic acid in the same syringe, since in the acidic medium the active principle of aminophylline - teufillin - precipitates.
Pharmacological interaction is divided into:
pharmacokinetic;
pharmacodynamic.
The pharmacokinetic interaction of drugs can occur at different levels:
Suction.
the effect of drugs on the pH in the gastrointestinal tract - the use of antacids leads to an increase in pH in the stomach, as a result of which the absorption of weak acid drugs and, therefore, the effects of these substances are reduced.
the effect of drugs on gastrointestinal motility - M-anticholinergic drugs and narcotic analgesics slow down the motility of the gastrointestinal tract, which causes longer contact of the drug with the mucosa and can lead to irritation (for example, when using aspirin).
Distribution.
Metabolism.
Excretion.
Pharmacodynamic interaction - this is the interaction of drugs, when one of them changes the process of generation and implementation of the pharmacological effect of the other. Pharmacodynamic interaction can occur in two ways:
Synergism.
Antagonism.
summarized - a simple addition of the effects of two or more drugs (for example, co-administration of two diuretics of ethacrylic acid and furosemide leads to the summation of their diuretic action), this type of interaction is expressed by the formula 1 + 1 \u003d 2;
potentiated - a type of interaction in which the pharmacological effect of a combination of drugs is greater than the mathematical sum of the pharmacological effects of each of the separately prescribed drugs (for example, with antipsychotic antipsychotics, droperidol significantly enhances the analgesic effect caused by opioid analgesic fentanyl); this type of drug interaction is expressed by the formula 1 + 1 \u003d 3.
Antagonism - this is the opposite effect of the drugs used simultaneously, when their overall pharmacological effect in the body is less than the sum of the effects of individual drugs. The following types of antagonism are:
physical, based on the physical interaction of substances (for example, activated carbon adsorbs toxins);
chemical, based on the chemical interaction of drugs (for example, with increased acidity, neutralization of hydrochloric acid in the stomach with antacid drugs);
competitive antagonism is observed when substances are similar in structure and compete for the same receptor (for example, the M-choline blocker atropine and the M-cholinomimetic pilocarpine compete for binding to M-cholinergic receptors);
non-competitive antagonism is observed with opposite effects of substances when acting on different receptors; noncompetitive antagonism may be functionalwhen substances act on different receptors of the same organ (for example, the excitatory effect of adrenaline and the inhibitory effect of acetylcholine on heart function) and physiologicalwhen substances act on different receptors of different organs (for example, aldosterone increases blood pressure by acting on the kidneys, and clonidine lowers blood pressure by acting on the central nervous system).
Pharmacology, its sections, tasks and place among medical, biological and specialized disciplines. Achievements of domestic pharmacology.
Pharmacology - biomedical science of medicinal substances and their effects on the body; in a broader sense, the science of physiologically active substances in general and their effect on biological systems.
Sections: General and private. General: 1) the principles of the production of medicines, their composition and properties 2) metabolism - pharmacokinetics and pharmacodynamics, 3) toxicology, 4) pharmacogenetics 5) pharmacogenomics.
Definition and structure of the recipe. Prescription Forms. General rules for prescribing. Features of prescribing toxic, narcotic, potent drugs.
Recipe- This is a doctor’s appeal to a pharmacist about the release of drugs to the patient, indicating the dosage form, dose and method of use. It is a medical, legal and monetary document in case of free or preferential dispensing of drugs. Doseexpressed in mass or volume units of the decimal system and is indicated in Arabic numerals. The number of whole grams is separated by a comma (1.0). More commonly used: 0.1 - one decigram; 0.01 - one centigram; 1.001 one milligram. The drops that make up the medicine are indicated by the Roman numeral, in front of which is written gtts. Biological units of action in the recipe thus indicate 500,000 units. Liquid substances in recipes are indicated in ml (0.1 ml). The recipe is certified by signature and personal seal. The prescription must indicate: the patient's age, date of prescription, last name and initials of the patient; surname and initials of the doctor, order of payment for the medicine. Moreover, preferential recipes are written out on special forms that have a stamp and seal. On special forms of another sample, drugs from the list of narcotic substances, sleeping pills, anorexigenic drugs are also prescribed. Moreover, the doctor prescribes the prescription, puts his signature and certifies it with a personal seal. In addition, it is signed by the head doctor or his deputy, the prescription has a round seal and a medical stamp
institutions. The same prescribing procedure is also defined for anabolic steroids, as well as phenobarbital, cyclodol, ephedrine hydrochloride, clonidine ( eye drops, ampoules), ointments of sunoref. On other forms of prescription forms, antipsychotics, tranquilizers, antidepressants, drugs containing ethyl alcohol, etc. are prescribed. For ambulatory patients it is forbidden to prescribe ether for anesthesia, chloroethyl, fentanyl, sombrevin, ketamine. The recipe begins with the word Recipe (Rp. - in abbreviated form), which means "take", then the names and quantities of written out are listed
medicinal substances in the genitive case. First called primary, then auxiliary. Next, the necessary dosage form is indicated. For instance, Misce ut fiat pulvis(M. f. pulvis) - "mix to make a powder." For dosed write: " Da tales doses numero10 "-" issue such doses with the number 10 ". At the end of the recipe after the word Signa(S) - "designate" in Russian (or national) language indicate the method of use of the medicine.
General Pharmacology, its sections. Examples common mechanisms the effects of drugs. The concept of medicine and poison.
1) the principles of the production of drugs, their composition and properties 2) metabolism - pharmacokinetics (the doctrine of absorption, distribution and biotransformation of them in the body) and Pharmacodynamics (the doctrine of the effect of drugs on the body) 3) toxicology, 4) pharmacogenetics (medical section genetics and pharmacology, studying the nature of the body's reactions to medicines depending on hereditary factors). 5) pharmacogenomics. (A branch of pharmaceuticals and pharmacology that studies the effect of each person’s genetic variation in his response to a drug)
When using drugs in the body, the latter can act on specific receptors, enzymes, cell membranes or directly interact with cell substances. The effect on specific receptors is based primarily on the fact that macromolecular structures are selectively sensitive to certain chemical compounds. The interaction of chemicals with the receptor is accompanied by physiological, biochemical changes in the body, which ultimately determine the clinical effect. Medicines - pharmacological agents (substances or mixtures of substances) that have undergone clinical trials and are approved for use for the prevention, diagnosis and treatment of diseases by an authorized body of the country in the prescribed manner, obtained from blood, blood plasma, as well as organs, tissues of humans or animals, plants, minerals, by synthesis or using biotechnology. Thus, medicinal products include substances of plant, animal or synthetic origin that have pharmacological activity and are intended for the production and manufacture dosage forms. Poison - a substance that leads in doses, even small relative to body weight, to disruption of the body: poisoning, intoxication, diseases and pathological conditions.
Pharmacodynamics
It studies the mechanism of action of drugs, as well as their biochemical and physiological effects. Her tasks include a description of the chemical and physical interactions between the drug and the target cell, as well as the full spectrum and severity of its pharmacological effects. Knowledge of pharmacodynamic patterns allows you to choose the right medication. Pharmacodynamic studies provide a deeper understanding of the regulation of biochemical and physiological processes in the body (Katzung B.G., 1998; Lawrence D.R. et al., 2002).
The action of most drugs is mediated by their binding to the macromolecules of the body. A change in the functional state of these macromolecules, in turn, triggers a chain of biochemical and physiological reactions that are converted into a pharmacological effect. Macromolecules with which chemicals interact are called receptors. Thus, any functionally active macromolecules can serve as receptors for drugs. Several important consequences follow from this statement. First, with the help of drugs, you can change the speed of any physiological process in the body. Secondly, drugs only change the natural physiological functions of the cell, without giving it new properties.
Receptors
Most receptors are proteins. These are receptors of hormones, growth factors, mediators, proteins involved in the most important metabolic and regulatory reactions (dihydrofolate reductase, acetylcholinesterase), transport proteins (Na +, K + -ATPase), structural proteins (tubulin). Cellular components of a different chemical nature, such as nucleic acids, with which antitumor agents interact, can also act as receptors.
Receptors of endogenous regulatory factors - hormones, mediators, etc., have pharmacological significance. These receptors serve as targets for many drugs, usually acting selectively due to the high specificity of the receptors for endogenous ligands. Medicines that, upon binding to the receptor, reproduce the physiological effect of the endogenous ligand, are called aganists, or stimulants. Drugs that do not cause this effect, but inhibit the binding of endogenous ligands, are called antagonists, or blockers. Substances whose effect is less pronounced than the effect of agonists are called partial agonists. Preparations stabilizing the receptor in an unactivated conformation are classified as inverse agonists.
Structural and functional dependence
The chemical structure of the drug rather rigidly determines its affinity for receptors and internal activity. A slight change in the chemical structure can significantly affect the pharmacological properties.
The synthesis of new drugs is largely based on this. Since chemical modification does not necessarily affect all pharmacological properties equally, it is possible to improve the effectiveness and safety of the drug, increase its selectivity, and improve the pharmacokinetic characteristics. For example, many hormone and mediator antagonists used in the clinic are synthesized by chemical modification of endogenous substances.
Drug Application Points
Since the effect of drugs is mediated by receptors, the point of application of the drug is determined not only by the features of its distribution, but also by the localization of the receptors, and the pharmacological effects depend on the functional significance of these receptors. The pharmacological effects of drugs whose receptors are common in many organs and tissues are diverse. If these receptors perform a function vital for cells, it is not only difficult to use the drug for therapeutic purposes, but it is also unsafe. Nevertheless, such drugs can be of great clinical importance. So, cardiac glycosides, widely used in heart failure, alter the transport of ions through the cell membrane, on which the vital activity of the cell depends. They have a narrow therapeutic range and are very toxic. Another example is antitumor agents. If the receptors with which the drug interacts are present on only a few types of differentiated cells, its effect is more selective. These drugs may have fewer adverse reactions, but still, these drugs may be toxic if their receptors perform a vital function. Some biological poisons (botulinum toxin, etc.) act in a similar way. In addition, even if the direct pharmacological effect is selective, its consequences may be more diverse.
Endogenous regulatory factor receptors
The term receptor refers to any macromolecular component of a cell with which a drug binds. One of the most important drug receptors is cellular proteins, which serve as receptors for endogenous regulatory factors - hormones, growth factors, mediators. By binding to the endogenous ligand, receptors transmit the signal from it into the target cell.
From the receptor, the signal arrives at the cellular targets (effector proteins) directly or through intermediate signaling molecules - protein-converters. The receptor, protein converters and effector proteins form the receptor-effector system. The closest effector protein in the signal transmission chain is often not a terminal effector (directly affecting cellular functions), but an enzyme or transport protein involved in the formation, transport, or inactivation of a second mediator — an ion or a small molecule. The second mediator, in turn, transfers information to a variety of intracellular targets, ensuring their simultaneous response to a signal from one receptor.
Receptors, converting proteins, and effector proteins not only transmit information. They also coordinate signals from different ligands, on the one hand, and all these signals with metabolic processes in the cell, on the other.
Acting as catalysts, receptors enhance the biological signal. Due to this important property, they serve as excellent targets for medicines. However, signal amplifiers are not only receptors with enzymatic activity, but all known receptors. Indeed, when a single ligand molecule binds to a receptor conjugated to an ion channel, many ions pass through the latter. The same is true for steroid hormone receptors: one hormone molecule triggers the transcription of many copies of mRNA, on the basis of which numerous protein molecules are synthesized.
The receptors of biologically active substances are divided into several classes depending on the structure and mechanism of action. The number of these classes is small.
Enzymatic receptors
The largest group of receptors with enzymatic activity are membrane receptors with their own protein kinase activity. They phosphorylate a variety of effector proteins located on the inside of the cell membrane. As a result, the function of these proteins or their interaction with other proteins changes.
There is another class of receptors with protein kinase activity - these are receptors conjugated with protein kinases. They lack an intracellular catalytic domain, but when interacting with an agonist, they bind or activate intracellular protein kinases on the inner surface of the membrane. These are receptors for neurotrophic factors and antigen-recognizing receptors for T and B lymphocytes consisting of several subunits. The latter also interact with phosphothyrosine phosphates. The function of other receptors that do not have an intracellular effector domain may be mediated by some other effector proteins.
Other receptors with their own enzymatic activity have a similar structure. These include, for example, receptors with their own phosphotyrosine phosphatase activity: their extracellular domain is similar in amino acid sequence to adhesion molecules. For many receptors with their own phosphotyrosine phosphatase activity, endogenous ligands are not known. However, according to genetic and biochemical studies conducted on different types of cells, the enzymatic activity of these receptors plays an important role. The intracellular domain of atrial natriuretic hormone receptors, other NUPs, as well as guaniline receptors, has its own guanylate cyclase activity and synthesizes cGMP, which acts as a second mediator. Perhaps there are other receptors with their own enzymatic activity.
Ion channel coupled receptors
The receptors of some mediators are directly associated with ion channels, when interacting with a ligand, selectively pass certain ions through the cell membrane (chemosensitive channels, ionotropic receptor channels, ionotropic receptors).
G-protein coupled receptors
This is a fairly large class of receptors that interact with effectors via G-proteins (proteins that use the substitution of guanine diphosphate (GDF) for guanine triphosphate (GTP). These include receptors for many biogenic amines, lipid signaling molecules (in particular eicosanoids), and various peptide and protein ligands. Enzymes (adenylate cyclase, phospholipase C) and potassium and calcium membrane channels act as effectors. The large number and important physiological role of receptors coupled to G-proteins makes them excellent. my targets for drugs: approximately half of all drugs prescribed by doctors (excluding antibiotics) act on these receptors.
A cell can carry up to 20 receptors on its surface, each of which selectively interacts with one or more types of G-proteins (differ in different types of α-subunits). The α-subunit is able to interact with one or more effector proteins, which allows you to coordinate signals from receptors of different ligands using one G-protein. On the other hand, a single receptor can trigger several mechanisms of intracellular signal transmission, activating several types of G-proteins, and act on different effector proteins through the same α-subunit. Such a complex system of divergence and convergence of signals provides flexible regulation of cellular functions (Ross, 1992).
Intracellular receptors
Receptors of steroid and thyroid hormones, calcitriol and retinoids are soluble intracellular DNA-binding proteins that regulate the transcription of certain genes (Mangelsdorf et al., 1994). These receptors belong to the superfamily of ligand-sensitive transcriptional regulators. The function of transcription factors is regulated by phosphorylation, interaction with cellular proteins, metabolites and other regulatory components of the cell.
Second Intermediary Systems
cAMP. Secondary intermediary systems are also involved in the integration of external signals. Although there are much more known receptors and protein signaling molecules than second mediators, the latter are involved in many pathways within the cell signal transmission. The most studied second intermediaries include cAMP, cGMP, Ca 2+, IF 3 (inositol triphosphate), DAG (diacylglycerol), NO. This group of heterogeneous compounds is constantly growing. Second mediators interact directly (by changing each other's metabolism) or indirectly (by acting on the same intracellular targets). The function of the second mediators, as well as the regulation of their formation (or release), cleavage and excretion from the cell, is conveniently considered with the example of cAMP. This second mediator is synthesized under the influence of adenylate cytase upon activation of many receptors conjugated with G-proteins. G s protein activates adenylate cyclase, G i protein inhibits.
There are at least 10 tissue-specific adenylate cyclotase isoforms that differ in the mechanisms of regulation of activity.
As a rule, cAMP activates protein kinases A (cAMP-dependent protein kinases), a small group of related proteins. These protein kinases, in turn, phosphorylate not only the final intracellular targets (enzymes, transport proteins), but also other protein kinases and other regulatory proteins. The latter include, for example, transcription factors. They are responsible for cAMP-mediated regulation of gene transcription, providing a delayed cellular response to the signal. In addition to the activation of protein kinases, cAMP acts directly on cationic membrane channels, which play an important role, in particular, in the functioning of neurons. Thus, the signal from cAMP causes a chain of biochemical changes in the target cell.
Calcium. Another well-studied second mediator is intracellular Ca 2+. Ca 2+ ions enter the cytoplasm in various ways: along the membrane channels (dependent on G-proteins, voltage-dependent, regulated by K + or Ca-Ca 2+), as well as through channels located in special areas of the endoplasmic reticulum and opening under the action of IF 3, and in skeletal muscle as a result of membrane depolarization. Removal of calcium from the cytosolic plasma occurs in two ways: it is absorbed by the endoplasmic reticulum or excreted from the cell. Ca 2+ transmits signals to a much larger number of proteins than cAMP - enzymes involved in cell metabolism, protein kinases, calcium-binding proteins. The latter interact with other final and intermediate effectors.
Receptor regulation
Receptors not only control physiological and biochemical functions, but also serve as objects of regulation. This regulation is carried out at the level of synthesis and decomposition of their macromolecules, through the formation of covalent bonds with other molecules, interaction with regulatory proteins, and receptor movement. Converting proteins and effector proteins are also subject to regulation. Regulatory signals can come from intracellular transmission pathways activated by stimulation of the receptor itself (via a feedback mechanism), as well as from other receptors (directly or indirectly).
Long-term stimulation of drug receptors usually leads to a decrease in reaction to it - at the same concentration, the drug causes a less pronounced effect. This phenomenon, called desensitization, refractoriness, and addiction, plays an important role in clinical practice: for example, with prolonged use of β-adrenergic agonists for the treatment of patients with AD, the severity of the reaction to these drugs decreases.
Homological desensitization applies only to stimulated receptors and is specific for the ligand. With heterologous desensitization, the severity of the reaction to other ligands, whose receptors act through the same intracellular signal transmission pathway, decreases. In the first case, negative feedback is provided by the effect on the receptor itself (phosphorylation, proteolysis, decreased synthesis), in the second case, in addition to the receptor, it can affect other proteins involved in intracellular signal transmission.
On the contrary, if receptors are not stimulated for a long time, their sensitivity to agonists increases (for example, with prolonged treatment with β-adrenoblocker propronolol, the sensitivity of β-adrenergic receptors to β-adrenostimulants increases).
Disorders due to impaired receptor function
In addition to individual differences in drug sensitivity, there are diseases caused by dysfunction of certain components of the mechanism of intracellular signal transmission from the receptor to the effector. With the loss of function of highly specialized receptors, the phenotypic manifestations of the disease may be limited (for example, with testicular feminization associated with a genetic absence or structural defects of androgen receptors). If a more universal mechanism inside the cell signal transmission is violated, the symptoms of the disease are more diverse, as, for example, with myasthenia gravis and some forms of insulin-resistant diabetes mellitus, caused respectively by autoimmune dysfunctions of N-cholinergic receptors and insulin receptors. Defects in any component involved in signal transduction from many receptors lead to multiple endocrine disorders. An example is the heterozygous form of G s protein deficiency that activates adenylate cyclase in all cells (Spiegel and Weinstein, 1995). A homozygous form of deficiency of this protein is likely to result in death.
Disturbances in the structure or localization of receptors can manifest as a weakened or enhanced reaction to the drug, as well as other undesirable effects.
Mutations encoding gene receptors are capable of changing both the response to a single use of the drug and the effectiveness of long-term treatment. For example, a defect in β-adrenergic receptors responsible for relaxing the smooth muscles of the bronchi and regulating airway resistance exacerbates the decrease in the sensitivity of these receptors to β-adrenostimulants during long-term treatment of patients with AD. As the mutations responsible for impaired receptor function are identified and the corresponding genes are cloned, it will be possible to develop methods for treating such diseases.
Receptor classification
Traditionally, drug receptors have been identified and classified based on the effects and relative activity of selective agonists (stimulants) and antagonists (blockers) acting on these receptors. For example, the effects of acetylcholine, which are reproduced when interacting with the cholinergic receptors of the muscarine alkaloid and are blocked by atropine, are called muscarinic effects, and the effects that are reproduced when interacting with the cholinergic receptors of nicotine are called nicotinic effects. Receptors that mediate the effects of muscarine and nicotine are called M and N cholinergic receptors, respectively. Although such a classification usually does not reflect the mechanism of action of drugs, it is convenient for systematizing their effects. Indeed, the assertion that a drug stimulates receptors of a certain type, at the same time determines the spectrum of effects of this drug and substances that enhance or weaken these effects. However, the validity of such claims can change with the identification of new types and subtypes of receptors, the discovery of additional mechanisms of action of drugs or previously unknown side effects.
Receptor Subtypes
With the advent of an ever-increasing variety of highly selective drugs, it has become clear that previously known types of receptors are divided into many subtypes. Molecular cloning methods have become a significant help in the study of new receptor subtypes, and the preparation of recombinant receptors has facilitated the creation of drugs that selectively act on these receptors. Different but related subtypes of receptors often (though not always) interact with different agonists and antagonists. Receptors for which no selective agonists or antagonists have been identified, usually do not belong to a single subtype, but to isoforms of the same receptor. Separate subtypes can also differ in the mechanisms of intracellular signal transmission. M 1 and M 3 cholinergic receptors, for example, act through the protein G q, which activates phospholipase C, indirectly causes the release of Ca 2+ from intracellular depots, and M 2 and M 4 cholinergic receptors through the protein G i, which inhibits adenylate cyclase. At the same time, the division of receptors into types and subtypes is often determined not by the mechanism of action, but by a random choice or is based on established ideas. So, α 1 -, α 2 - and β-adrenergic receptors differ in response to drugs and in signal transmission (activate proteins G i, G q and G s, respectively), although α and β-adrenergic receptors are of different types, and α 1 - and α 2 -adrenoreceptors - to different subtypes within the same type. Isoforms of α 1 -adrenoreceptors α 1A, α 1B and α 1D differ little in their biochemical properties; the same is characteristic of isoformrase subtypes of β-adrenergic receptors (β 1, β 2 and β 3).
Differences between receptor subtypes are used to create highly selective drugs, for example, drugs that have different effects on the same tissue due to binding to receptor subtypes that differ in the mechanisms of intracellular signal transmission. In addition, drugs can selectively target certain cells or tissues expressing receptors of a subtype. The greater the selectivity of drugs (in relation to a certain tissue or in relation to a certain effect), the more favorable is the ratio of its benefits and undesirable effects.
Using molecular genetic methods, not only different isoforms of receptors have been discovered, but also genes encoding new, previously unknown receptors. Many of these receptors are already assigned to one or another known class, and their function has been studied using the corresponding ligands. However, ligands have not yet been found for some receptors.
The discovery of many isoforms of the same receptor encoded by different genes (especially if the isoforms do not differ in the mechanisms of intracellular signal transmission and interact with the same endogenous ligands) allows the expression of receptors in different cells to be independently regulated in accordance with the needs of the body in different age periods.
Non-receptor mediated drug action
Not all drugs act through the macromolecular structures - receptors. Some drugs interact with small molecules or ions that are present in the body normally or in one or another pathological condition. So, antacids neutralize hydrochloric acid in the stomach. Mesna (a drug that is quickly excreted by the kidneys and neutralizes free radicals) binds to the active metabolites of some antitumor drugs, reducing the severity of adverse reactions from the urinary tract. A number of biologically inactive substances (for example mannitol) can be introduced in quantities sufficient to increase the osmolarity of biological fluids, and thus change the distribution of extracellular and intracellular fluids. With the help of these substances it is possible to increase diuresis, increase bcc, eliminate cerebral edema. In addition, they are used as laxatives.
Some drugs can integrate into the components of the cell and change their functions due to structural similarities with the substances that make up these components. For example, analogues of purines and pyrimidines are inserted into nucleic acids and are used as antiviral and antitumor agents.
A.P. Viktorov "Clinical Pharmacology"