Mechanisms of action medicinal substances - these are the ways in which substances cause pharmacological effects. The main mechanisms of action of drugs include:
Physical.
The mechanism of direct chemical interaction.
Membrane (physico-chemical).
Enzymatic (biochemical).
Receptor.
The physical mechanism of action. The action of a drug substance is associated with its physical properties. For example, activated carbon is specially processed, and therefore has a high surface activity. This allows him to absorb gases, alkaloids, toxins, etc.
Direct chemical interaction. This is a rather rare mechanism of action of drugs, the essence of which is that drugs directly interact with molecules or ions in the body. Such a mechanism of action is possessed, for example, by the unitiol drug belonging to the group of antidotes. In case of poisoning with thiol poisons, including salts of heavy metals, unitiol enters into a direct chemical reaction with them, resulting in the formation of non-toxic complexes that are excreted in the urine. Thus, antacids also act, which enter into a direct chemical interaction with hydrochloric acid, lowering the acidity of gastric juice.
Membrane (physicochemical) mechanism. It is associated with the effect of drugs on ion currents (Na +, K +, Cl - and others), which determine the transmembrane electric potential. According to this mechanism, anesthetics, antiarrhythmic drugs, local anesthetics, etc.
Enzymatic (biochemical) mechanism. This mechanism is determined by the ability of some drugs to exert an activating or inhibitory effect on enzymes. The arsenal of drugs with such a mechanism of action is very wide. For example, anticholinesterase drugs, monoamine oxidase inhibitors, proton pump blockers, etc.
Receptor mechanism. In the human body there are highly specific biologically active substances (mediators) that interact with receptors and change the functions of various organs or tissues of the body.
Receptors are macromolecular structures with selective sensitivity to certain chemical compounds. With the interaction of drugs with receptors, biochemical and physiological changes in the body occur, accompanied by one or another clinical effect.
Mediators and drugs that activate receptors and cause a biological effect are called agonists. Medicinal substances that bind to receptors, but do not cause their activation and biological effect, reduce or eliminate the effects of agonists, are called antagonists. Allocate also antagonist agonists - substances that act differently on subtypes of the same receptors: they stimulate some receptor subtypes, and block others. For example, the narcotic analgesic nalbuphine stimulates opioid kappa receptors (therefore, reduces pain sensitivity) and blocks opioid mu receptors (therefore, is less dangerous in terms of drug dependence).
The ability of substances to bind to receptors is referred to by the term “affinity”. In relation to the same receptors, the affinity of different substances can be different.
The following types of receptors are distinguished:
Plasma membrane receptors:
channel type: N-type cholinergic receptors, muscle-type H-cholinergic receptors, GABA receptors;
g-protein receptors: α- and β-adrenergic receptors, M 3 -cholinoreceptors;
integrative type receptors: NO receptor.
Cytosolic.
Mitochondrial.
Pharmacodynamics is a section general pharmacologystudying the features of the action of drugs on the body. Namely, pharmacodynamics studies:
- mechanisms of action of drugs;
- end pharmacological effects;
- dependence of the action of drugs on various conditions;
- the effects of drugs upon repeated administration;
- combined action of drugs;
- drug incompatibility;
- side effects of drugs.
Mechanisms of action medicines
The mechanisms of action of drugs are the ways in which substances cause pharmacological effects. The main mechanisms of action of drugs include:
- Physical.
- The mechanism of direct chemical interaction.
- Membrane (physico-chemical).
- Enzymatic (biochemical).
- Receptor.
The physical mechanism of action. The action of a drug substance is associated with its physical properties. For example, activated carbon is specially processed, and therefore has a high surface activity. This allows him to absorb gases, alkaloids, toxins, etc.
Direct chemical interaction. This is a rather rare mechanism of action of drugs, the essence of which is that drugs directly interact with molecules or ions in the body. Such a mechanism of action is possessed, for example, by the unitiol drug belonging to the group of antidotes. In case of poisoning with thiol poisons, including salts of heavy metals, unitiol enters into a direct chemical reaction with them, resulting in the formation of non-toxic complexes that are excreted in the urine. Thus, antacids also act, which enter into a direct chemical interaction with hydrochloric acid, lowering the acidity of gastric juice.
Membrane (physicochemical) mechanism. It is associated with the effect of drugs on ion currents (Na +, K +, Cl - and others), which determine the transmembrane electric potential. According to this mechanism, anesthetics, antiarrhythmic drugs, local anesthetics, etc.
Enzymatic (biochemical) mechanism. This mechanism is determined by the ability of some drugs to exert an activating or inhibitory effect on enzymes. The arsenal of drugs with such a mechanism of action is very wide. For example, anticholinesterase drugs, monoamine oxidase inhibitors, proton pump blockers, etc.
Receptor mechanism. In the human body there are highly specific biologically active substances (mediators) that interact with receptors and change the functions of various organs or tissues of the body.
Receptors are macromolecular structures with selective sensitivity to certain chemical compounds. With the interaction of drugs with receptors, biochemical and physiological changes in the body occur, accompanied by one or another clinical effect.
Mediators and drugs that activate receptors and cause a biological effect are called agonists. Medicinal substances that bind to receptors, but do not cause their activation and biological effect, reduce or eliminate the effects of agonists, are called antagonists. Allocate also antagonist agonists - substances that act differently on subtypes of the same receptors: they stimulate some receptor subtypes, and block others. For example, the narcotic analgesic nalbuphine stimulates opioid kappa receptors (therefore, reduces pain sensitivity) and blocks opioid mu receptors (therefore, is less dangerous in terms of drug dependence).
The ability of substances to bind to receptors is referred to by the term “affinity”. In relation to the same receptors, the affinity of different substances can be different.
The following types of receptors are distinguished:
- Plasma membrane receptors:
- channel type: N-type cholinergic receptors, muscle-type H-cholinergic receptors, GABA receptors;
- g-protein receptors: α- and β-adrenergic receptors, M 3 -cholinoreceptors;
- integrative type receptors: NO receptor.
- Cytosolic.
- Mitochondrial.
- Nuclear
Plasma membrane receptors.
Channel Type Receptors
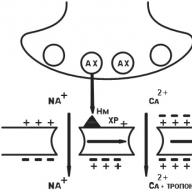
N n -cholinergic receptor of the nerve type (CNS, autonomic ganglia, sinocarotid zone, chromaffin adrenal gland tissue). After the binding of acetylcholine (AX) with H n -cholinergic receptors, Na + channels open and Na rushes into the cell, bearing a positive charge. The postsynaptic membrane is depolarized. There is an action potential that shifts along the membrane of the neuron, opening up electrically dependent Na + channels. A nerve impulse arises in the postganglionic fiber (Fig. 6).
Fig. 6. N n -cholinergic receptor
N m - muscle type cholinergic receptor (skeletal muscle cell membranes). The initial processes are similar, but electrically dependent Ca ++ channels open. Ca ++ ions enter the muscle fiber, Ca ++ is released from the sarcoplasmic reticulum. The level of Ca ++ rises, which induces muscle contraction (Fig. 7).

Fig. 7. N m-cholinergic receptor
GABA receptors. These are receptors for γ-aminobutyric acid (GABA). GABA interacts with GABA receptors, in the structure of which there are chloride channels. As a result of receptor stimulation, channels open and chlorine ions (Cl -) freely enter the cell. An increase in the concentration of chlorine ions inside the cell leads to hyperpolarization of the membrane and a decrease in the activity of neurons. It is more difficult to excite such a cell (Fig. 8).

Fig. 8. GABA receptor:
GABA-R - GABA-receptor, BD-R - benzodiazepine receptor, BR - barbiturate receptor
Receptors associated with G protein
G-proteins, i.e., GTP-binding (guanosine triphosphate-binding) proteins, are localized in the cell membrane and consist of α-, β- and γ-subunits. They (G-proteins) regulate the activity of specific effectors (instant messengers, secondary intermediaries). These messengers can be enzma (adenylate cyclase, phospholipase); channels for potassium, calcium, sodium; some transport proteins. Each cell can have many G-proteins, each of them regulates the activity of various messengers, while changing the function of the cell.
M 3 -cholinergic receptor (smooth muscle membranes (MMCs) and exocrine gland cells). Acetylcholine stimulates M 3 -XR bound to the G protein. Phospholipase-C (FLS) is activated, which catalyzes the cleavage of PIDP (phosphatidylinositol diphosphate) into ITP (inositol triphosphate) and DAG (diacylglycerol). ITF entering the cytoplasm of MMC releases Ca ++ from cavelles .

Fig. 9. M3-cholinergic receptor
Ca ++ binds to calmodulin, activates myosine kinase (MK), which catalyzes the phosphorylation of myosin light chains, which leads to cell contraction (Fig. 9). Similarly, an impulse is transmitted at the synapses of the secretory glands.
Norepinephrine stimulates α 1 -adrenoreceptorBy starting the following chain of events:
Norepinephrine (HA) → α 1 -adrenoreceptor → activation of the α-subunit of G s-protein → activation of PLL → FIDF cleavage → increase in ITF concentration → increase in Ca 2+ concentration in the cell → Ca 2+ binds to calmodulin → myosine kinase is activated → light chains phosphorylate → light chains myosin → myosin interacts with actin → a reduction in MMC develops (Fig. 10).

Fig. 10. α 1 -adrenoreceptor
b 1 -receptor(fig. 11). Norepinephrine → activates b 1 -AP → activation of the α-subunit of G-protein → activation of AC → increase in cAMP production from ATP → increase in cAMP concentration in cardiomyocyte → activation of protein kinases → phosphorylation of calcium channel proteins → increase in Ca 2+ entry through channels and increase Ca concentration 2+ in the cell → an increase in the force of contractions of the heart.

Fig. eleven. b 1 receptor
b 2 -receptor(fig. 12). ON → b 2 -AP → activation of the α-subunit of G-protein → activation of AC → increased cAMP formation → stimulated protein kinase → kinase that catalyzes the phosphorylation of myosine kinase is cleaved, while the latter’s activity is lost → myosin phosphorylation does not occur → MMC relaxation.
Regulation of the release of HA from nerve endings is carried out by the neurotransmitter itself upon excitation of the α 2 -AP presynaptic membrane. The emission of HA decreases.

Fig. 12. b 2 receptor
Integrative type receptors
These are receptors that are proteins that penetrate the membrane. In this case, the outer part of the protein plays a receptor role, while the inner part plays a catalytic role (Fig. 13).

Fig. thirteen. Integrative type receptor
Cytosolic receptors
Under physiological conditions, such receptors serve to bind steroid hormones (sex hormones, glucocorticoids). These substances enter the cell and bind to cytosolic receptors there. This complex penetrates the nucleus and changes the work of the genome there. As a result, protein synthesis in the cell changes (Fig. 14).
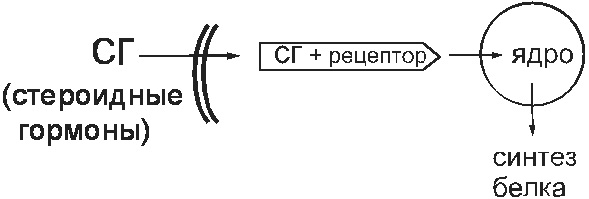
Fig. 14. Cytosolic receptor
Mitochondrial receptors
In the mitochondria there are also receptors with which medicinal substances interact, such as triiodothyronine hydrochloride, which are analogues of the natural hormone T 3. As a result of this interaction, ATP synthesis increases.
Nuclear receptors
T 3 penetrates the nucleus and there interacts with receptors of this type. As a result, the work of the genome changes and new proteins are synthesized.
Final pharmacological effects (according to Vershinin)
Despite the abundance of drugs, the changes caused by them in the body are of the same type (Fig. 15). The effect of any drug on the organs can be reduced to five main pharmacological effects (according to N.V. Vershinin):
- Soothing - reduction to normal of increased organ function (use of sedatives).
- Oppression - decrease below the norm of the function of the body (use of drugs for anesthesia).
- Paralysis - cessation of reduced organ function (respiratory depression in case of an overdose of narcotic analgesics).
- Toning up - strengthening of reduced function to normal (use of β 1 -adrenomimetics).
- Excitation - Increasing the function of the organ in excess of the norm (the use of diuretics in case of poisoning, expectorant drugs).
Fig. 15. Final pharmacological effects

Types of action of drugs
- The main thing is secondary.
the main thing an action is that which underlies the therapeutic or prophylactic administration of a drug. Collateral - undesirable, dangerous for the patient action of drugs.
- Reversible, irreversible.
Once in the body, medicinal substances interact with those cells that have a biological substrate capable of reacting with this substance. This interaction depends on the chemical structure of the drug. The binding of a drug substance to an appropriate substrate is reversible if they (substrate and drug) bind to each other for a while.
In few cases, the therapeutic goal requires irreversible turning off a structure from its function. This applies, for example, to most antimicrobial, antitumor agents that are able to form strong (covalent) bonds with the elements of DNA helix cells ("crosslinking") or bacterial enzymes, as a result of which the cells lose their ability to reproduce.
- Direct, indirect (indirect).
Direct the action implies that the therapeutic effect is due to the direct interaction of the drug with the biosubstrate of the diseased organ and directly leads to certain shifts. If the function of the organ (system) changes a second time as a result direct influence drug on another organ, another system, this action is called indirect (indirect). Cardiac glycosides improve myocardial contractility ( direct action) and, as a result, improve blood circulation in the body, which is accompanied by an improvement in diuresis (indirect effect).
A special case of an indirect action is reflex act. For example, vasodilation and improvement of trophic tissue as a result of irritation of the ends of the sensory nerves of the skin.
- Selective, non-selective.
Selective action is the effect of therapeutic doses of drugs on specific receptors. For example, the effect of salbutamol on β 2 -adrenoreceptors. It should be borne in mind that the selectivity of drugs is relative, with increasing doses, it disappears.
- Local, resorptive.
Local the effect of the drug is carried out before it is absorbed into the blood (for example, ointment).
Resorptive (systemic) action develops after absorption of the drug into the blood. The vast majority of drugs have this effect.
In the vast majority of cases, for a drug substance (ligand) to exert its effect, it must meet specific components in the body - target receptors, molecular structures that are protein, less often nucleic acids, lipid or other configurations located inside or on the surface of cells, with which it interacts, starting a chain of biochemical and physico-chemical processes leading to a certain effect.
There are two types of membrane receptors - ion channels and receptors associated with the G-protein. For example, the sodium channel is characteristic of adetylcholine and similar drugs. Acetylcholine interacts with channel protein, causing conformational changes in it, which contribute to the opening of the channel and the penetration of sodium ions into the cell. This process underlies nervous excitement. Some medicinal substances, interacting with the protein of the sodium channel, prevent its opening, thereby blocking the transmission of nerve excitation.
The so-called G-protein is attached to the inner part of the plasma membrane of cells, which ensures the synchronization of the drug substance interaction process with the simultaneous activation of the corresponding intracellular target proteins. As shown in the figure, the drug molecule interacts with the receptor (P) on the outer surface of the membrane, which causes conformational changes in the receptor protein. Due to this, the G-protein changes its spatial structure, migrates in the plane of the membrane to enzymes that are in an inactive state inside the cell. The interaction of G-protein with enzymes (T) determines their activation (LV / P / T). Norepinephrine, dopamine and other ligands interact specifically with receptors associated with G-protein. It should be noted that acetylcholine can interact not only with the channel protein, but also with receptors associated with the G-protein.
For the interaction between the ligand and the bioreceptor to occur, it is necessary that they have a complementarity, that is, between them there must be a certain affinity, or affinity (correspondence of size, spatial configuration, the presence of opposite charges, etc.). For example, a negative charge of a receptor must correspond to a positive charge of an exogenous ligand, and nonpolar radicals of a substance can bind to hydrophobic sites of a receptor.
Among the physicochemical properties of medicinal substances that affect their interaction with receptors, we should single out the size of the molecule, depending on which the substance can interact with the entire receptor or with its constituent. The kinetics of its penetration through biological membranes also depends on the size of the drug molecule. Typically, as the size of a molecule increases, its flexibility and the possibility of the formation of van der Waals bonds with a macromolecular partner increase. In addition, the stereochemistry of a drug molecule is important. The pharmacological activity depends on the isomeric form of the drug substance. And one must keep in mind: the stiffer the conformation of the receptor molecule, the greater the difference in the action of stereoisomers.
The interaction of the drug substance - the receptor is due to intermolecular bonds. Initially, a substance is attracted to the receptor by electrostatic forces, and in the presence of complementarity, it forms bonds with the receptor using physical and physicochemical interactions (typical for drugs that are excreted from the body in an unchanged or unchanged form) or chemical interactions (inherent in compounds that undergo chemical transformations in the body). The weakest van der Waals forces take part in determining the specificity of the interaction of a drug substance with biochemical reactive systems. Hydrogen bonds are involved in the processes of recognition and fixation of a substance (ligand) to biostructures. Ionic bonds arise in cases where medicinal substances contain a cationic or anionic group, and the opposite structures are in bioreceptors. Often, ionic bonds form at the first stages of a pharmacological reaction between substances and receptors. In such cases, the effect of the drug is reversible. The formation of coordination covalent bonds is important. With their participation, interactions of alkylating agents with biosubstrates, as well as drugs and antidotes with metals, occur during the formation of stable chelate complexes, for example, unithiol with arsenic or tetacin-calcium with lead. The action of such substances is irreversible.
In addition, there is a hydrophobic interaction. Although the energy of its bonds is small, the interaction of a large number of long aliphatic chains leads to the appearance of stable systems. Hydrophobic interactions play a role in stabilizing biopolymer conformations and forming biological membranes.
Amino acid residues in the protein receptor molecule contain polar and non-polar groups that determine the formation of polar and non-polar bonds between them and drug substances. The polar groups (-OH, -NH, COO-, -N 3 H, \u003d O) provide the formation of mainly ionic and hydrogen bonds. Nonpolar groups (hydrogen, methyl, cyclic radicals, etc.) form hydrophobic bonds with low molecular weight medicinal substances.
Thus, the interaction of drugs with specific receptors can be achieved through various chemical bonds having unequal strength. So, the approximate strength of curare-like substances with cholinergic receptors for electrostatic (ionic) interaction is 5 kcal / mol, dipole-ion - 2-5 kcal / mol, dipole-dipole - 1-3 kcal / mol, hydrogen bonds - 2-5 kcal / mol, van der Waals bonds — 0.5 kcal / mol, hydrophobic bonds — 0.7 kcal per CH 2 group. The decrease in bond strength depending on the distance between atoms for electrostatic interaction is r -2, the dipole ion is r -3, the dipole dipole is r -4, the hydrogen bonds are r -4, the van der Waals bonds are r -7 . This kind of connection can be broken, which ensures the reversibility of the action of drugs. More durable are covalent bonds, which provide a long and often irreversible effect of substances, for example, alkylating antitumor drugs. Most drugs bind reversibly to receptors. In this case, as a rule, the nature of the compound is very complex: ionic, dipole-dipole, van der Waals, hydrophobic, and other types of bonds can simultaneously participate in it, which is largely determined by the complementary nature of the substance and receptor and, accordingly, the degree of their convergence between by myself.
The binding strength of a substance to receptors is denoted by the term "affinity". Substances acting on the same receptors may have a different degree of affinity for them. In this case, substances with a higher affinity can displace substances with a lower affinity from the compound with receptors. To determine the equilibrium state between the "occupied" receptors (DR), free receptors and free substance (D), the dissociation constant (K D) is used, which is determined by the following formula:
K D \u003d [D] * [R] / [DR]
The negative logarithm of K D (pR D) is an indicator of affinity. To characterize affinity, the pD 2 indicator is often used, i.e., the negative logarithm of EC 50, (the concentration of the substance in which it causes an effect of 50% of the maximum effect).
The variety of chemical interaction bonds and their unequal strength, or affinity between ligands and bioreceptors, is explained by the complex structure of drugs containing radicals with different reactivity and having a multidimensional volumetric shape, as well as the complexity of the interaction processes, which often occur in several stages (phases): complex formation drug substance is a receptor; intramolecular grouping; complex dissociation.
Thus, only substances with a pronounced affinity for the bioreceptor can cause a pharmacological effect. The severity of the effect depends on the concentration of the drug and the total number of receptors.
If substances have sufficient internal activity, then they are called agonists. By internal activity is understood the ability of agonists to cause a biological effect by changing the conformation of receptors, i.e., the ability of a ligand to activate a receptor. This phenomenon is considered as an affinity of the agonist-receptor complex to the transducer; the conversion of external signals into internal ones is called transduction. Intracellular signal transmission underlies processes such as contraction of muscle fibers, cell division, proliferation, differentiation, etc. It has now been established that the cell has many substances (hormones, bioactive peptides, nucleotides, steroids, low molecular weight bioregulators, etc.) specific receptors. As a result of the interaction of these substances with these specific receptors, secondary messengers (intermediaries) are formed that trigger a cascade of biochemical reactions.
There is a concept of " partial agonists"- medicinal substances that, when bound to receptors, do not give the maximum effect. This incomprehensible phenomenon is supposedly due to the incomplete (lesser) dependence of the affinity of the drug-receptor complex for the traneductor. For example, a partial opiate receptor agonist nalorphine acts similarly to the full agonist of these receptors morphine, although weaker than the latter. At the same time, when used together, nalorphine weakens or eliminates the effects of morphine; in particular, the inhibitory effect of morphine on respiration is eliminated. Isoprenaline is a true agonist, and prenalterol is a partial agonist for β-adrenergic receptors. According to receptor theory, a true agonist can induce a maximum response, even if it interacts with only part of the receptors.
Specific receptors may have the same or different binding sites for agonists and antagonists. Different binding sites for different agonists are possible. In the case where the agonist and antagonist have the same binding sites and the blocking effect of the antagonist on the receptor is completely eliminated by increasing the concentration of the agonist (the maximum effect of the agonist is achieved), the relationship between the antagonist and the agonist is designated as competitive antagonism. If the binding sites for the agonist and antagonist are different, then the relationship between them is defined as non-competitive antagonism. To characterize the antagonists, pA 2 is often used (the negative logarithm of the molar concentration of the antagonist, at which its concentration must be doubled to obtain the standard effect of the agonist).
Under the conditions of a whole organism, agonists and antagonists cause changes in certain physiological functions. In this case, the action of antagonists is determined by the fact that they inhibit the influence of specific natural ligands on specific receptors (for example, the atropine M-cholinergic antagonist inhibits the action of their acetylcholine agonist). Changes that are directly related to the interaction of substances with specific receptors are designated by the term "primary pharmacological reaction, which may be the beginning of a series of reactions leading to the stimulation or inhibition of certain physiological functions."
Changes in the functions of organs or systems (for example, changes in the strength and frequency of heart contractions, smooth muscle tone of internal organs, secretion of glands, blood pressure, etc.) caused by a drug substance are designated as pharmacological effects of this substance. So, for cardiac glycosides, the primary pharmacological reaction is inhibition of the activity of the transport Na +, K-ATPase of the myocardial fibers, which is regarded as a possible specific receptor for cardiac glycosides. In this regard, K + entry into muscle fibers and exit from Na + fibers are disrupted, and the Ca2 + content in the cytoplasm increases, which promotes the interaction of actin and myosin. The result of these changes is an increase in heart rate, which is the main pharmacological effect of cardiac glycosides.
The prolonged exposure of specific receptors to agonists is often accompanied by a decrease in their sensitivity. The latter may be associated with a change in receptors, a decrease in their number (density), or a disruption in the processes that follow the excitation of receptors. In this case, the pharmacological effects of agonists become less pronounced.
Thus, the pharmacological effects of most drugs are related to their effect on the corresponding specific receptors.
Substances with high affinity for the bioreceptor and low internal activity are called antagonists or blockers, since they, without causing changes in the conformation of the bioreceptor, interfere with the interaction of endogenous and / or exogenous agonist ligands with it. There are so-called "secondary or dumb receptors with which medicinal substances bind, but have no pharmacological effect. Such "dumb" receptors are most often present in proteins and blood plasma (but can also be found in tissues). Connection with "mute" receptors leads to a decrease in the concentration of free drug substance, and therefore to a decrease in the therapeutic effect.
Numerous modern theories explaining the mechanism of ligand-receptor interaction, the state of the receptors themselves, the lack of proportionality between the number of occupied receptors and the final reaction, changes in signal transmission efficiency and the existence of reserve receptors and partial agonists, etc., formed the basis for ideas about the mechanism of action of representatives of various groups of drugs. These interactions are divided into receptor interaction and chemical interaction.
The mechanism of interaction of drugs with bioreceptor can be depicted as the following scheme: each ligand (drug substance or physiological substrate) binds to a specific site on a particular receptor. Activated receptors directly or indirectly regulate ion fluxes (1) and / or other intracellular processes (secretion or muscle contraction) or activate the guanine nucleotide-binding proteins (G-proteins) system, which, in turn, enhances the activation of the second enzyme-mediator system. Several different second mediators function in the cytoplasm, activating various target proteins, for example, protein kinases. The latter act on specific substrates and mediate the pharmacological effect.
From the presented description it can be seen that the action of drugs is carried out by the following mechanisms:
- physiological functions of the tissue (for example, contractile, secretory) can be regulated by several receptors, and therefore various ligands;
- between the interaction of the drug substance with the receptor and the response of the tissue or organ, there may be several intermediate steps, in particular the activation of second mediator-related systems of the receptors;
- the effectiveness of the mechanisms responsible for the stimulus-response sequence, as well as the density of receptors, can vary from tissue to tissue.
The therapeutic effect of certain drugs is due to their direct (unrelated to specific receptors) chemical interaction with endogenous compounds or other mechanisms of interaction (osmotic pressure, adsorption). So for osmotic diuretics - mannitol, urea - there are no specific receptors. These substances increase the osmotic pressure in the renal tubules, as a result of which the reabsorption of water is disturbed and diuresis increases. The action of adsorbing substances, acid-forming diuretics is not associated with specific receptors.
Antacids (e.g. aluminum or magnesium hydroxides) react with hydrochloric acid to form products with weak acid properties. Chelating agents, binding to some metals, form inactive chemical complexes.
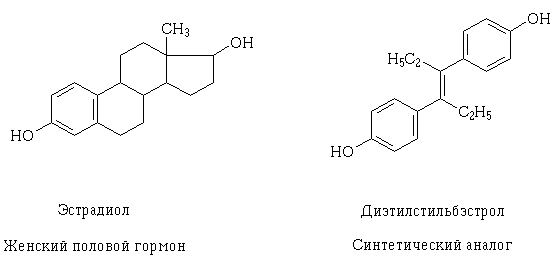
As knowledge on the structure of receptors and the mechanism of the possible pharmacodynamic interaction of drugs at the cellular level deepens, it is possible to purposefully create them, as well as explain why drugs may have such an effect, which differ at first glance in their structure. An example of such a phenomenon is estradiol and the transisomer of diethylstilbestrol, a synthetic analogue of the female genital. Their structural molecules are different, but contain the same hydroxy groups in properties and sizes, similarly located and oriented in space, due to which the molecules of these substances can interact with the same receptor and have a similar pharmacological effect.
The ways in which medicinal substances cause certain pharmacological effects are denoted by the term "mechanisms of action." This concept is used to explain the effects of drugs at the molecular, organ and systemic levels. For example, the mechanism of action of anticholinesterase agents at the molecular level is reduced to the blockade of acetylcholinesterase by interacting with its anionic and esterase centers. At the same time, explaining the mechanism of the hypotensive effect of anticholinesterase drugs, bradycardia and vasodilation are indicated as the cause of this effect, that is, they consider the mechanism of this effect at the organ level.
Studies of the mechanisms of action of drugs are ongoing, and ideas about the mechanism of action of a drug substance as new data are obtained can not only become more detailed, but also significantly change.