Overwhelming majority medicines is having therapeutic effect by changing the activity of the physiological systems of cells that are produced in the body during evolution. Under the influence of a medicinal substance in the body, as a rule, a new type of cell activity does not arise, only the rate of various natural processes changes. Inhibition or excitation of physiological processes leads to a decrease or enhancement of the corresponding functions of body tissues.
Medicines can act on specific receptors, enzymes, cell membranes or directly interact with cell substances. Details of the mechanisms of action medicinal substances studied in a course of general or experimental pharmacology. Below we provide only some examples of the main mechanisms of action of drugs.
Effect on specific receptors. Receptors are macromolecular structures that are selectively sensitive to certain chemical compounds. The interaction of chemicals with the receptor leads to the occurrence of biochemical and physiological changes in the body, which are expressed in a particular clinical effect.
Drugs that directly excite or increase the functional activity of receptors are called agonists, and substances that inhibit the action of specific agonists are called antagonists. Antagonism can be competitive and non-competitive. In the first case, the drug substance competes with a natural regulator (mediator) for binding sites in specific receptors. A receptor blockade caused by a competitive antagonist can be eliminated with large doses of an agonist or natural mediator.
A variety of receptors are divided by sensitivity to natural mediators and their antagonists. For example, acetylcholine-sensitive receptors are called cholinergic, adrenaline-sensitive receptors are called adrenergic. In terms of sensitivity to muscarine and nicotine, cholinergic receptors are divided into muscarinic-sensitive (m-cholinergic receptors) and nicotine-sensitive (n-cholinergic receptors). H-cholinergic receptors are heterogeneous. It is established that their difference lies in sensitivity to various substances. There are n-cholinergic receptors located in the ganglia of the autonomic nervous system, and n-cholinergic receptors of the striated muscle. Various subtypes of adrenergic receptors are known, denoted by the Greek letters α1, α 2, β1, β2.
H1 and H2 histamine, dopamine, serotonin, opioid and other receptors are also isolated.
Effect on enzyme activity. Some drugs increase or inhibit the activity of specific enzymes. For example, physostigmine and neostigmine reduce the activity of cholinesterase, which destroys acetylcholine, and give effects characteristic of excitation of the parasympathetic nervous system. Monoamine oxidase inhibitors (iprazide, nialamide), which prevent the destruction of adrenaline, increase the activity of the sympathetic nervous system. Phenobarbital and zixorin, increasing the activity of liver glucuronyl transferase, reduce the level of bilirubin in the blood.
Physico-chemical effect on cell membranes. The activity of cells of the nervous and muscle systems depends on the flow of ions that determine the transmembrane electrical potential. Some drugs alter ion transport.
So antiarrhythmic, anticonvulsant drugs, drugs for general anesthesia.
Direct chemical interaction. Medicines can directly interact with small molecules or ions inside the cells. For example, ethylenediaminetetraacetic acid (EDTA) strongly binds lead ions. The principle of direct chemical interaction underlies the use of many antidotes for chemical poisoning. Another example is the neutralization of hydrochloric acid with antacids.
Pharmacodynamics
It studies the mechanism of action of drugs, as well as their biochemical and physiological effects. Her tasks include a description of the chemical and physical interactions between the drug and the target cell, as well as the full spectrum and severity of its pharmacological effects. Knowledge of pharmacodynamic patterns allows you to choose the right medication. Pharmacodynamic studies provide a deeper understanding of the regulation of biochemical and physiological processes in the body (Katzung B.G., 1998; Lawrence D.R. et al., 2002).
The action of most drugs is mediated by their binding to the macromolecules of the body. A change in the functional state of these macromolecules, in turn, triggers a chain of biochemical and physiological reactions that are converted into a pharmacological effect. Macromolecules with which chemicals interact are called receptors. Thus, any functionally active macromolecules can serve as receptors for drugs. Several important consequences follow from this statement. First, with the help of drugs, you can change the speed of any physiological process in the body. Secondly, drugs only change the natural physiological functions of the cell, without giving it new properties.
Receptors
Most receptors are proteins. These are receptors of hormones, growth factors, mediators, proteins involved in the most important metabolic and regulatory reactions (dihydrofolate reductase, acetylcholinesterase), transport proteins (Na +, K + -ATPase), structural proteins (tubulin). Cellular components of a different chemical nature, such as nucleic acids, with which antitumor agents interact, can also act as receptors.
Receptors of endogenous regulatory factors - hormones, mediators, etc., have pharmacological significance. These receptors serve as targets for many drugs, usually acting selectively due to the high specificity of the receptors for endogenous ligands. Medicines that, upon binding to the receptor, reproduce the physiological effect of the endogenous ligand, are called aganists, or stimulants. Drugs that do not cause this effect, but inhibit the binding of endogenous ligands, are called antagonists, or blockers. Substances whose effect is less pronounced than the effect of agonists are called partial agonists. Preparations stabilizing the receptor in an unactivated conformation are classified as inverse agonists.
Structural and functional dependence
The chemical structure of the drug rather rigidly determines its affinity for receptors and internal activity. A slight change in the chemical structure can significantly affect the pharmacological properties.
The synthesis of new drugs is largely based on this. Since chemical modification does not necessarily affect all pharmacological properties equally, it is possible to improve the effectiveness and safety of the drug, increase its selectivity, and improve the pharmacokinetic characteristics. For example, many hormone and mediator antagonists used in the clinic are synthesized by chemical modification of endogenous substances.
Drug Application Points
Since the effect of drugs is mediated by receptors, the point of application of the drug is determined not only by the features of its distribution, but also by the localization of the receptors, and the pharmacological effects depend on the functional significance of these receptors. The pharmacological effects of drugs whose receptors are common in many organs and tissues are diverse. If these receptors perform a function vital for cells, it is not only difficult to use the drug for therapeutic purposes, but it is also unsafe. Nevertheless, such drugs can be of great clinical importance. So, cardiac glycosides, widely used in heart failure, alter the transport of ions through the cell membrane, on which the vital activity of the cell depends. They have a narrow therapeutic range and are very toxic. Another example is antitumor agents. If the receptors with which the drug interacts are present on only a few types of differentiated cells, its effect is more selective. These drugs may have fewer adverse reactions, but still, these drugs may be toxic if their receptors perform a vital function. Some biological poisons (botulinum toxin, etc.) act in a similar way. In addition, even if the direct pharmacological effect is selective, its consequences may be more diverse.
Endogenous regulatory factor receptors
The term receptor refers to any macromolecular component of a cell with which a drug binds. One of the most important drug receptors is cellular proteins, which serve as receptors for endogenous regulatory factors - hormones, growth factors, mediators. By binding to the endogenous ligand, receptors transmit the signal from it into the target cell.
From the receptor, the signal arrives at the cellular targets (effector proteins) directly or through intermediate signaling molecules - protein-converters. The receptor, protein converters and effector proteins form the receptor-effector system. The closest effector protein in the signal transmission chain is often not a terminal effector (directly affecting cellular functions), but an enzyme or transport protein involved in the formation, transport, or inactivation of a second mediator — an ion or a small molecule. The second mediator, in turn, transfers information to a variety of intracellular targets, ensuring their simultaneous response to a signal from one receptor.
Receptors, converting proteins, and effector proteins not only transmit information. They also coordinate signals from different ligands, on the one hand, and all these signals with metabolic processes in the cell, on the other.
Acting as catalysts, receptors enhance the biological signal. Due to this important property, they serve as excellent targets for medicines. However, signal amplifiers are not only receptors with enzymatic activity, but all known receptors. Indeed, when a single ligand molecule binds to a receptor conjugated to an ion channel, many ions pass through the latter. The same is true for steroid hormone receptors: one hormone molecule triggers the transcription of many copies of mRNA, on the basis of which numerous protein molecules are synthesized.
Depending on the structure and mechanism of action, the receptors of biologically active substances are divided into several classes. The number of these classes is small.
Enzymatic receptors
The largest group of receptors with enzymatic activity are membrane receptors with their own protein kinase activity. They phosphorylate a variety of effector proteins located on the inside of the cell membrane. As a result, the function of these proteins or their interaction with other proteins changes.
There is another class of receptors with protein kinase activity - these are receptors conjugated with protein kinases. They lack an intracellular catalytic domain, but when interacting with an agonist, they bind or activate intracellular protein kinases on the inner surface of the membrane. These are receptors for neurotrophic factors and antigen-recognizing receptors for T and B lymphocytes consisting of several subunits. The latter also interact with phosphothyrosine phosphates. The function of other receptors that do not have an intracellular effector domain may be mediated by some other effector proteins.
Other receptors with their own enzymatic activity have a similar structure. These include, for example, receptors with their own phosphotyrosine phosphatase activity: their extracellular domain is similar in amino acid sequence to adhesion molecules. For many receptors with their own phosphotyrosine phosphatase activity, endogenous ligands are not known. However, according to genetic and biochemical studies conducted on different types of cells, the enzymatic activity of these receptors plays an important role. The intracellular domain of atrial natriuretic hormone receptors, other NUPs, as well as guaniline receptors has its own guanylate cyclase activity and synthesizes cGMP, which acts as a second mediator. Perhaps there are other receptors with their own enzymatic activity.
Ion channel coupled receptors
The receptors of some mediators are directly associated with ion channels, when interacting with a ligand, selectively pass certain ions through the cell membrane (chemosensitive channels, ionotropic receptor channels, ionotropic receptors).
G-protein coupled receptors
This is a fairly large class of receptors that interact with effectors via G-proteins (proteins that use the substitution of guanine diphosphate (GDF) for guanine triphosphate (GTP). These include receptors for many biogenic amines, lipid signaling molecules (in particular eicosanoids), and various peptide and protein ligands. Enzymes (adenylate cyclase, phospholipase C) and potassium and calcium membrane channels act as effectors. The large number and important physiological role of receptors coupled to G-proteins makes them excellent. my targets for drugs: approximately half of all drugs prescribed by doctors (excluding antibiotics) act on these receptors.
A cell can carry up to 20 receptors on its surface, each of which selectively interacts with one or more types of G-proteins (differ in different types of α-subunits). The α-subunit is able to interact with one or more effector proteins, which allows you to coordinate signals from receptors of different ligands using one G-protein. On the other hand, a single receptor can trigger several mechanisms of intracellular signal transmission, activating several types of G-proteins, and act on different effector proteins through the same α-subunit. Such a complex system of divergence and convergence of signals provides flexible regulation of cellular functions (Ross, 1992).
Intracellular receptors
Receptors of steroid and thyroid hormones, calcitriol and retinoids are soluble intracellular DNA-binding proteins that regulate the transcription of certain genes (Mangelsdorf et al., 1994). These receptors belong to the superfamily of ligand-sensitive transcriptional regulators. The function of transcription factors is regulated by phosphorylation, interaction with cellular proteins, metabolites and other regulatory components of the cell.
Second Intermediary Systems
cAMP. Secondary intermediary systems are also involved in the integration of external signals. Although there are much more known receptors and protein signaling molecules than second mediators, the latter are involved in many pathways within the cell signal transmission. The most studied second intermediaries include cAMP, cGMP, Ca 2+, IF 3 (inositol triphosphate), DAG (diacylglycerol), NO. This group of heterogeneous compounds is constantly growing. Second mediators interact directly (by changing each other's metabolism) or indirectly (by acting on the same intracellular targets). The function of the second mediators, as well as the regulation of their formation (or release), cleavage and excretion from the cell, is conveniently considered with the example of cAMP. This second mediator is synthesized under the influence of adenylate cytase upon activation of many receptors conjugated with G-proteins. G s protein activates adenylate cyclase, G i protein inhibits.
There are at least 10 tissue-specific adenylate cyclotase isoforms that differ in the mechanisms of regulation of activity.
As a rule, cAMP activates protein kinases A (cAMP-dependent protein kinases), a small group of related proteins. These protein kinases, in turn, phosphorylate not only the final intracellular targets (enzymes, transport proteins), but also other protein kinases and other regulatory proteins. The latter include, for example, transcription factors. They are responsible for cAMP-mediated regulation of gene transcription, providing a delayed cellular response to the signal. In addition to the activation of protein kinases, cAMP acts directly on cationic membrane channels, which play an important role, in particular, in the functioning of neurons. Thus, the signal from cAMP causes a chain of biochemical changes in the target cell.
Calcium. Another well-studied second mediator is intracellular Ca 2+. Ca 2+ ions enter the cytoplasm in various ways: along the membrane channels (dependent on G-proteins, voltage-dependent, regulated by K + or Ca-Ca 2+), as well as through channels located in special areas of the endoplasmic reticulum and opening under the action of IF 3, and in skeletal muscle as a result of membrane depolarization. Removal of calcium from the cytosolic plasma occurs in two ways: it is absorbed by the endoplasmic reticulum or excreted from the cell. Ca 2+ transmits signals to a much larger number of proteins than cAMP - enzymes involved in cell metabolism, protein kinases, calcium-binding proteins. The latter interact with other final and intermediate effectors.
Receptor regulation
Receptors not only control physiological and biochemical functions, but also serve as objects of regulation. This regulation is carried out at the level of synthesis and decomposition of their macromolecules, through the formation of covalent bonds with other molecules, interaction with regulatory proteins, and receptor movement. Converting proteins and effector proteins are also subject to regulation. Regulatory signals can come from intracellular transmission pathways activated by stimulation of the receptor itself (via a feedback mechanism), as well as from other receptors (directly or indirectly).
Long-term stimulation of drug receptors usually leads to a decrease in reaction to it - at the same concentration, the drug causes a less pronounced effect. This phenomenon, called desensitization, refractory, addictive, plays an important role in clinical practice: for example, with prolonged use β-adrenergic agonists for the treatment of patients with AD the severity of the reaction to these drugs is reduced.
Homological desensitization applies only to stimulated receptors and is specific for the ligand. With heterologous desensitization, the severity of the reaction to other ligands, whose receptors act through the same intracellular signal transmission pathway, decreases. In the first case, negative feedback is provided by the effect on the receptor itself (phosphorylation, proteolysis, decreased synthesis), in the second case, in addition to the receptor, it can affect other proteins involved in intracellular signal transmission.
On the contrary, if receptors are not stimulated for a long time, their sensitivity to agonists increases (for example, with prolonged treatment with β-adrenoblocker propronolol, the sensitivity of β-adrenergic receptors to β-adrenostimulants increases).
Disorders due to impaired receptor function
In addition to individual differences in drug sensitivity, there are diseases caused by dysfunction of certain components of the mechanism of intracellular signal transmission from the receptor to the effector. With the loss of function of highly specialized receptors, the phenotypic manifestations of the disease may be limited (for example, with testicular feminization associated with a genetic absence or structural defects of androgen receptors). If a more universal mechanism inside the cell signal transmission is violated, the symptoms of the disease are more diverse, as, for example, with myasthenia gravis and some forms of insulin-resistant diabetes mellitus, caused respectively by autoimmune dysfunctions of N-cholinergic receptors and insulin receptors. Defects in any component involved in signal transduction from many receptors lead to multiple endocrine disorders. An example is the heterozygous form of G s protein deficiency that activates adenylate cyclase in all cells (Spiegel and Weinstein, 1995). A homozygous form of deficiency of this protein is likely to result in death.
Disturbances in the structure or localization of receptors can manifest as a weakened or enhanced reaction to the drug, as well as other undesirable effects.
Mutations encoding gene receptors are capable of changing both the response to a single use of the drug and the effectiveness of long-term treatment. For example, a defect in β-adrenergic receptors responsible for relaxing the smooth muscles of the bronchi and regulating airway resistance exacerbates the decrease in the sensitivity of these receptors to β-adrenostimulants during long-term treatment of patients with AD. As the mutations responsible for impaired receptor function are identified and the corresponding genes are cloned, it will be possible to develop methods for treating such diseases.
Receptor classification
Traditionally, drug receptors have been identified and classified based on the effects and relative activity of selective agonists (stimulants) and antagonists (blockers) acting on these receptors. For example, the effects of acetylcholine, which are reproduced when interacting with the cholinergic receptors of the muscarine alkaloid and are blocked by atropine, are called muscarinic effects, and the effects that are reproduced when interacting with the cholinergic receptors of nicotine are called nicotinic effects. Receptors that mediate the effects of muscarine and nicotine are called M and N cholinergic receptors, respectively. Although such a classification usually does not reflect the mechanism of action of drugs, it is convenient for systematizing their effects. Indeed, the assertion that a drug stimulates receptors of a certain type, at the same time determines the spectrum of effects of this drug and substances that enhance or weaken these effects. However, the validity of such claims can change with the identification of new types and subtypes of receptors, the discovery of additional mechanisms of action of drugs or previously unknown side effects.
Receptor Subtypes
With the advent of an ever-increasing variety of highly selective drugs, it has become clear that previously known types of receptors are divided into many subtypes. Molecular cloning methods have become a significant help in the study of new receptor subtypes, and the preparation of recombinant receptors has facilitated the creation of drugs that selectively act on these receptors. Different but related subtypes of receptors often (though not always) interact with different agonists and antagonists. Receptors for which no selective agonists or antagonists have been identified, usually do not belong to a single subtype, but to isoforms of the same receptor. Separate subtypes can also differ in the mechanisms of intracellular signal transmission. M 1 and M 3 cholinergic receptors, for example, act through the protein G q, which activates phospholipase C, indirectly causes the release of Ca 2+ from intracellular depots, and M 2 and M 4 cholinergic receptors through the protein G i, which inhibits adenylate cyclase. At the same time, the division of receptors into types and subtypes is often determined not by the mechanism of action, but by a random choice or is based on established ideas. So, α 1 -, α 2 - and β-adrenergic receptors differ in response to drugs and in signal transmission (activate proteins G i, G q and G s, respectively), although α and β-adrenergic receptors are of different types, and α 1 - and α 2 -adrenoreceptors - to different subtypes within the same type. Isoforms of α 1 -adrenoreceptors α 1A, α 1B and α 1D differ little in their biochemical properties; the same is characteristic of isoformrase subtypes of β-adrenergic receptors (β 1, β 2 and β 3).
Differences between receptor subtypes are used to create highly selective drugs, for example, drugs that have different effects on the same tissue due to binding to receptor subtypes that differ in the mechanisms of intracellular signal transmission. In addition, drugs can selectively target certain cells or tissues expressing receptors of a subtype. The greater the selectivity of drugs (in relation to a certain tissue or in relation to a certain effect), the more favorable is the ratio of its benefits and undesirable effects.
Using molecular genetic methods, not only different isoforms of receptors have been discovered, but also genes encoding new, previously unknown receptors. Many of these receptors are already assigned to one or another known class, and their function has been studied using the corresponding ligands. However, ligands have not yet been found for some receptors.
The discovery of many isoforms of the same receptor encoded by different genes (especially if the isoforms do not differ in the mechanisms of intracellular signal transmission and interact with the same endogenous ligands) allows the expression of receptors in different cells to be independently regulated in accordance with the needs of the body in different age periods.
Non-receptor mediated drug action
Not all drugs act through the macromolecular structures - receptors. Some drugs interact with small molecules or ions that are present in the body normally or in one or another pathological condition. So, antacids neutralize hydrochloric acid in the stomach. Mesna (a drug that is rapidly excreted by the kidneys and neutralizes free radicals) binds to the active metabolites of some antitumor drugs, reducing the severity of adverse reactions from the urinary tract. A number of biologically inactive substances (for example mannitol) can be introduced in quantities sufficient to increase the osmolarity of biological fluids, and thus change the distribution of extracellular and intracellular fluids. With the help of these substances it is possible to increase diuresis, increase bcc, eliminate cerebral edema. In addition, they are used as laxatives.
Some drugs can integrate into the components of the cell and change their functions due to structural similarities with the substances that make up these components. For example, analogues of purines and pyrimidines are inserted into nucleic acids and are used as antiviral and antitumor agents.
A.P. Viktorov "Clinical Pharmacology"
As a rule, the mechanism of action of drugs is based on their ability to initiate (trigger) complex biochemical n / or biophysical processes that ultimately alter and / or optimize the functional activity of the target cell.
Medicines can carry out their action against organs and / or target cells by:
Direct chemical interaction;
Physico-chemical interaction on the cell membrane;
Actions on specialized enzymes;
Actions on regulatory genes;
Actions on specific receptors.
Direct chemical interaction LS. This mechanism of action of drugs is quite rare and can be realized outside the cell, for example, in the lumen of the stomach or intestines. Its essence lies in the fact that drugs enter into a direct chemical reaction with molecules and / or ions that are formed in the body in a normal state when a pathological condition occurs. An example of direct chemical interaction is the chemical reaction of neutralization of hydrochloric acid of the stomach when taking antacid drugs (see T. 2, p. 112).
Physico-chemical interaction of drugs on the cell membrane. One of the main functions of the cytoplasmic membrane is the implementation of ion exchange between the cytoplasm and the extracellular environment. Transmembrane ion exchange can also take place through special voltage-dependent transmembrane ion channels - sodium, potassium, calcium, chlorine, etc. Some drugs, reaching the cell membrane, interact with these channels and alter their functional activity. So, for example, the antiarrhythmic effect of a class IA drug, quinidine, is based on its ability to block the passage of Na + ions through transmembrane sodium channels (see T. 2, p. 35).
The effect of drugs on specialized enzymes. A relatively small amount of drugs realizes its pharmacological effect by changing the activity of some specialized cellular enzymes. Medicines that increase the activity of cellular enzymes are called enzyme inducers. Such action is possessed, for example, by sleeping pills and anticonvulsant drug phenobarbital, which significantly enhances the activity of microsomal liver enzymes. The biological significance of this effect of phenobarbital and close to it LS will be considered below.
Medicines that inhibit the activity of specialized enzymes are called enzyme inhibitors. So, for example, the antidepressant from the group of monoamine oxidase inhibitors (MAOs), the drug pirlindole realizes its antidepressant effect by inhibiting the activity of the MAO enzyme in the central nervous system (see T. 1, p. 294).
The ability to inhibit the activity of the enzyme acetylcholinesterase is the basis of the pharmacological activity of anticholinesterase drugs, for example physostigmine. It is known that under physiological conditions, acetylcholinesterase inactivates (destroys) acetylcholine, a neurotransmitter that transmits excitation in the synapses of the parasympathetic nervous system. Physostigmine, suppressing the activity of acetylcholinesterase, promotes the accumulation in the synapses of the parasympathetic system of the neurotransmitter acetylcholine, as a result of which the tone of the parasympathetic nervous system increases, which is manifested at the systemic level by the development of bradycardia, lowering blood pressure (BP), and increasing motility of the gastrointestinal tract (GIT), pupil, etc.
Medicines can interact reversibly and irreversibly with enzymes. For example, the drug enalapril reversibly inhibits the activity of the angiotensin converting enzyme, which entails, in particular, a decrease in blood pressure, while organophosphorus toxic substances irreversibly inhibit the activity of acetylcholinesterase.
The effect of drugs on regulatory genes. Currently, scientists are making attempts to create drugs that realize their pharmacological effects by directly affecting the physiological activity of regulatory genes. This trend seems especially promising after the structure of the human genome was deciphered in 2000. It is believed that the selective normalization of the function of regulatory genes under the influence of drugs will make it possible to achieve success in the treatment of many, including previously incurable, diseases.
The effect of drugs on receptors. Before moving on to the specifics of the interaction of drugs with receptors, it is necessary to clarify what we mean by the term “receptor” (from Latin recipio - take, take).
From the course of physiology it is known that the term "receptor" means highly specialized formations that are able to perceive, transform and transmit the energy of an external signal to the nervous system. Such receptors are called sensory (from lat. Sensus - feeling, sensation, perception).
Sensory receptors include receptors of the organs of hearing, vision, smell, taste, touch, etc. Sensory receptors of these organs belong to the so-called exteroreceptors.
If the presence of sensory organs that respond to external stimuli of irritation has been known since ancient times, then the presence of sensory receptors inside the body was questioned until the middle of the 19th century. For the first time, the presence of such receptors inside the body was suggested by the Russian physiologist I.F.Pion, who showed in 1866 a drop in blood pressure due to aortic irritation in a rabbit experiment. This discovery gave rise to the search and study of receptors located inside the body, and these receptors themselves were called interoreceptors.
By the beginning of the 20th century a sufficient number of sensory interoreceptors was revealed and their important role in the regulation of the physiological functions of the body was proved.
In 1905, J. Langley proved that when a drug is applied to a cell membrane, a pharmacological effect develops if it is applied only to a specific area of \u200b\u200bit. Moreover, this site makes up only a small part of the total area of \u200b\u200bthe cell surface. This observation allowed J. Langley to conclude that specialized receptor sites interacting with drugs exist on the cell membrane.
However, the priority in creating the receptor theory of the action of drugs belongs to the German physiologist P. Ehrlich, who in 1906 first introduced the term “receptor” and formulated the postulate “the drug does not work if it is not fixed on the cell membrane”. According to the theory of P. Ehrlich, a drug molecule has two functionally active groups, one of which ensures its fixation on the cell surface in the region of the drug receptor, and the second functional group interacts with the receptor and triggers a complex chain of biochemical reactions that alter its (cell) physiological activity .
Thus, as early as the beginning of the 20th century. it became obvious that there are at least two classes of interoreceptors: sensory receptors that transmit information about the state of internal organs and body tissues to the central nervous system; labeling receptors that interact with drugs that alter the functional activity of target cells.
It should immediately be noted that in the future, in the text of the textbook, in order to avoid confusion in terminology, receptors for drugs and biologically active substances, i.e. labeled, or cytoreceptors. will be denoted by the term “receptor”, while sensory interoreceptors will be denoted by a term characterizing their functional activity, for example, “baroreceptors”, “pain receptors”, etc.
The discovery by P. Ehrlich on the cell membrane of drugs receptors served as a starting point for the development of pharmacological science, in particular pharmacodynamics, one of the main tasks of which is to study the receptor mechanisms of action of drugs.
At present, the structure of a large number of cellular receptors, the features of the interaction of certain biologically active compounds with them have been revealed, which made it possible, on the one hand, to understand the mechanism of action of known drugs, and on the other hand, was the basis for creating new highly effective drugs.
Naturally, it is difficult to imagine that in the course of evolution, receptors for various synthetic (chemically obtained) drugs were formed in the human body, especially since the vast majority of drugs presented on the modern pharmaceutical market have been synthesized in the last 50 years or less. It is proved that the receptor apparatus of the cell is a very ancient functional-structural formation. So, a- and β-adrenergic receptors (receptors, the interaction of which norepinephrine and adrenaline affect the functional activity of the cell) are found not only in animal cells, but also on the cell membranes of plant cells, for example, in the cells of the plant nittella, where a- and β- adrenorecentors regulate the movement of protoplasm (cell content).
Then what are the receptors for drugs discovered by P. Ehrlich, and why do they interact with them?
Currently, there is no doubt that the so-called drug receptors are actually receptors for endogenous (produced in the body) biologically active substances involved in the regulation of the functional activity of internal organs and body tissues. Such biologically active compounds include substances released from the nerve endings at the time of transmission of the nerve signal, as well as hormones, vitamins, amino acids, etc. For each endogenous biologically active substance, there are strictly specific receptors for it. So, for example, the biologically active substance produced in the body, adrenaline, can activate strictly specific a- and β-adrenoreceptors, and glucocorticosteroids - hormones of the adrenal cortex - interact only with glucocorticosteroid receptors strictly specific to them.
Synthetic drugs that realize their effects by interacting with the receptor apparatus of the cell, in their chemical structure, are more or less similar to endogenous biologically active compounds that interact with similar receptors. For example, synthetic vasoconstrictor (causing vasoconstriction) drugs phenylephrine is close in its chemical structure to the endogenous biologically active substance norepinephrine, therefore, like norepinephrine, it has the ability to stimulate a-adrenoreceptors.
Sometimes, due to the peculiarities of their chemical structure, drugs can interact not with the receptor itself, but with the adjacent portion of the cell membrane. Since in this case, the drug does not interact with the receptor itself, but with the adjacent portion of the cell membrane, they speak not of an exciting or blocking effect on the receptor, but of an allosteric (from the Greek alios - another, different) effect, or effect. As a result, a change in both the structure of the membrane adjacent to the receptor and individual components of the receptor itself can occur, which may entail a change in the sensitivity of the receptor to a biologically active substance specific to it. In cases where the sensitivity of the receptor to a biologically active substance increases, they speak of sensitization (from Latin sensus - sense) or sensitization (from Latin sensibilis - sensitivity) of the receptor, and in cases where the sensitivity of the receptor decreases, they speak of desensitization receptor.
The peculiarity of the allosteric effect lies in the fact that drugs that have this kind of mechanism of action do not directly affect the transmission of a nerve impulse, but modify it in the desired direction. For example, the mechanism of action of anxiolytics (anti-anxiety drugs; synonym: tranquilizers), which in their chemical structure are derivatives of benzodiazepine, is based on the phenomenon of allosteric excitation of postsynaptic benzodiazepine receptors. The excitation of the latter, in turn, promotes the activation of inhibitory postsynaptic receptors of gamma-aminobutyric acid (GABA receptors), which is clinically manifested by the elimination of symptoms of neurotic diseases such as anxiety, anxiety, fear, etc.
Receptors, interacting with which a biologically active substance or drug in any way changes the functional state of a target cell, are called specific.
In addition to specific receptors, so-called drug-specific receptors are isolated. In the specialized medical literature, these receptors are also called the "place of loss" of drugs. By contacting such receptors, drugs do not have any biological effect, but they themselves become biologically inactive. An example of this type of receptor can serve as receptors located on plasma proteins, in particular, on water-soluble proteins - albumin. The significance of this phenomenon will be discussed in detail below (see T. 1, p. 72).
The structure of the receptors is quite complex, but most of them are protein macromolecules or glycoproteins, which may also include ions, lipids, nucleic acids, etc. Receptor i.e. the protein macromolecule forming it is characterized by a specific, specific for each receptor, spatial arrangement of its chemical groups. The protein macromolecule forming the receptor can be integrated (immersed) in the lipid bilayer of the cytoplasmic membrane or localized inside the cell. The main function of a cell receptor is to “recognize” a chemical signal transmitted to it through an endogenous biologically active substance and / or drugs and transform it into the corresponding biochemical and / or biophysical response of the cell.
It was previously believed that drugs or endogenous biologically active substances interact with receptors of the “key and lock” type, i.e. the receptor has such a structure that allows the drug to find “your” receptor, connect to it and, as it were, “turn on” and “turn it off”. However, with the development of medical science, it has become apparent that the ego is not quite so. At present, the molecular processes of the conversion of extracellular signals into intracellular, regulating cell function, have already been studied quite well. mechanisms that result in the effect of the interaction of endogenous biologically active substances or drugs with receptors.
When interacting with the receptor of an endogenous biologically active substance and / or an active L C like it, a conformation occurs - a spatial change in the form of a protein macromolecule, which is the trigger for various intracellular processes that determine the response of a target cell to a mediator and / or drug. For example, activation of bronchial smooth muscle adrenergic receptors under the influence of the β 2 -adrenostimulator phenoterol leads to an increase in the activity of the enzyme adenylate cyclase, which promotes the accumulation of cyclic adenosine monophosphate (cAMP) in the cell and, as a result, cell relaxation.
In general biological terms, cellular receptors can be considered as strictly specialized “sensory organs” of cells, through which they perceive “information” emanating, for example, from the central nervous system and / or endocrine system. Despite the important role of the receptor apparatus, receptors occupy only an insignificant part of the cell membrane. For example, the M-cholinergic receptor apparatus of a cell occupies no more than 1/6 000 of its surface area.
Studying the characteristics of the interaction of drugs with the receptor, on the one hand, allows us to understand the basis of the molecular mechanism of its action, and on the other hand, provides information on what changes should be made in the structure of drugs to enhance its ability to interact with this receptor, i.e. . allows targeted synthesis of new highly effective drugs.
Under physiological conditions, different cellular receptors do not function independently, but are in constant interaction with each other, thereby regulating the specific activity of the cell. For example, activation of cardiac β-adrenoreceptors by endogenous norepinephrine causes, in particular, an increase in the number of heart contractions, and activation of cardiac M-cholinergic receptors by endogenous acetylcholine, on the contrary, causes a decrease in the number of heart contractions.
A great contribution to the understanding of the receptor mechanisms of action of drugs was made by the discovery of pre- and postsynaptic receptors. Synapse (from Greek synapsis - connection, connection) is a specialized contact zone between nerve cells or other excitable structures of the body, which ensures the transmission of incoming information and the preservation of its informational significance. The study of the structure and functional role of synapses was begun at the end of the 19th century. after that, the Spanish histologist S. Ramon n Cajal (S. Ramon at Cajal) suggested the presence of a specialized transmission system in the central nervous system. Synapses got their name in 1897, when the English physiologist C. Sherrington proposed this term to refer to the contact area between nerve cells.
Currently, there are three types of synapses:
1) “electrical” synapses in which information is transmitted by transferring an electrical signal from a pre-synaptic membrane. This type of synapse is called efaps (from the Greek. Ephapsis - tight contact);
2) "chemical" synapses in which information is transmitted through special biologically active substances - neurotransmitters (from the Greek. Neuron - nerve and Latin. Mediator - mediator; synonym: mediator);
3) “mixed” synapses in which information is transmitted both chemically and electrically.
The pharmacological effects of the vast majority of drugs that affect the functions of synapses are realized by their effect on the goth or another stage of signal transmission in chemical synapses, i.e. in synapses of the second kind.
As a rule, chemical synapses are classified by neurotransmitters that transmit nerve impulses in them, as follows:
Synapses in which acetylcholine acts as a mediator are called cholinergic;
Synapses in which adrenaline and norepinephrine act as a mediator are called adrenergic;
Synapses in which ATP and adenosine act as a mediator are called purinergic;
Synapses in which gamma-aminobutyric acid acts as a mediator are called GABA-ergic, etc.
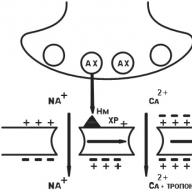
The structure of the synapse is currently well understood. The synapse consists of a presynaptic process of a nerve cell (axon end) and a “signal”-receiving apparatus located on the membrane of an effector (“executive”) cell.
The axon of the efferent neuron, approaching the effector cell, loses the myelin sheath, expands and forms the so-called presynaptic thickening (Fig. 1.5). The surface of the nerve ending facing the cell membrane of the effector cell is called the presynaptic membrane. The site of the effector cell opposite the presynaptic membrane is called the postsynaptic membrane (see Fig. 1.5). Depending on the structural features of the synapse, the presynaptic membrane may have more or less folds and thus has a larger or smaller area. In chemical synapses, the presynaptic membrane does not directly contact the postsynaptic membrane, but is separated from it by a small distance called the synaptic cleft (see Fig. 1.5).
Presynaptic thickening, i.e. the end portion of the axon contains a larger number of mitochondria, intracellular organelles involved in the synthesis and accumulation of energy, which is greater than the body of the neuron, which indicates the intensity of the energy processes that occur in this section of the nerve cell. In addition to mitochondria, presynaptic thickenings contain a large number of small vesicles - vesicles. On average, about 20,000 vesicles are contained in one presynaptic thickening. The latter are located unevenly in the presynaptic thickening, as a rule, most of them are located near the presynaptic membrane. The neurotransmitter is synthesized in the body and axon of the neuron and accumulates in the vesicles. Each vesicle contains several thousand molecules of a neurotransmitter (from I 000 to 50 000). When a nerve impulse occurs, the vesicle fuses with the presynaptic membrane and the neurotransmitter is secreted into the synaptic cleft (see Fig. 1.5).
Fig. 1.5. Schematic diagram of the structure of the "chemical" synapse:
a is a schematic image; b - electronic micrograph; 1- presynaptic nerve ending; 2 - prssynaptic membrane; 3 - postsynaptic membrane; 4 - synaptic cleft; B - vesicle; NM - neurotransmitter; P - postsynaptic receptor: OZ - "reverse" capture of a neurotransmitter; SF is a specialized enzyme that destroys excess neurotransmitter in the synaptic cleft
Functionally active receptor formations are located on the postsynaptic membrane, which are able to interact with the neurotransmitter released from the presynaptic membrane during the passage of a nerve impulse. Receptors located on the postsynaptic membrane are called synaptic or postsynaptic receptors in specialized medical literature. By postsynaptic receptors is meant macromolecules of protein nature embedded in the postsynaptic membrane with genetically predetermined structure and function, capable of reversibly interacting with neurotransmitters and / or drugs due to the functional groups of the active center (the “recognizing” part of the macromolecule).
The transmission of the nerve signal in the synapse occurs as follows: under the influence of a nerve stimulus, the vesicles move to the presynaptic membrane and the neurotransmitter is secreted by the exocytosis into the synaptic cleft (see Fig. 1.5). The neurotransmitter released into the synaptic cleft reaches the postsynaptic membrane, where, interacting with the postsynaptic receptor, it triggers a chain of biochemical and / or biophysical reactions, the result of which is the physiological response of the target cell. However, not all the amount of released neurotransmitter reaches postsynaptic receptors and interacts with them. Part of the neurotransmitter is captured by the presynaptic membrane and “returns” to storage sites. This phenomenon is called the neurotransmitter reuptake phenomenon.
The remaining amount of non-interacting neurotransmitter receptor is destroyed in the synaptic cleft by specialized enzymes. This phenomenon is called the degradation of neurotransmitters. For example, the enzyme acetylcholinesterase catalyzes (accelerates) the process of degradation (destruction) in the synaptic cleft of the neurotransmitter acetylcholine.
Unlike the neurotransmitter, its metabolic products have neurotransmitter activity. The whole process of interaction of the neurotransmitter with receptors and the destruction of its excess by a specific enzyme is extremely short and does not exceed 2 ms (1 ms \u003d 0.001 s).
Such a short duration of this process is explained, on the one hand, by the extremely rapid release of the neurotransmitter from the receptor, and on the other hand, by the high rate of enzymatic inactivation of the neurotransmitter in the synaptic shel.
The fundamentally functional activity of the synapse can be changed as follows:
To accelerate, reduce or block the synthesis, accumulation and / or catabolism (destruction) of the neurotransmitter in the presynaptic ending. As a result of this, the content of the neurotransmitter and, as a consequence of this, the intensity of its physiological activity will somehow change.
For example, the sympatholytic reserpine prevents the accumulation of catecholamines in synaptic vesicles until their complete emptying. As a result, the amount of the neurotransmitter norepinephrine released into the synaptic cleft drops sharply. At the system level, this effect is realized in the form of a decrease in blood pressure. Some drugs do not directly affect the content of neurotransmitters in the presynaptic ending, but inhibit the activity of enzymes that destroy them. Thus, a number of antidepressants act. For example, the antidepressant pirlindol inhibits (suppresses) the activity of the monoamine oxidase enzyme at the presynaptic end and, as a result, increases the concentration of neurotransmitters such as norepinephrine, dopamine, and serotonin in it. Clinically, this effect of pirlindol is manifested by a decrease in feelings of anxiety and fear, improved mood, increased physical activity, etc .;
Change (facilitate, complicate) the ability of the neurotransmitter to penetrate the presynaptic membrane and, therefore, increase or decrease the amount of the neurotransmitter released into the synaptic cleft with each pulse.
For example, the psychostimulant amphetamine facilitates the release of catecholamines in the adrenergic synapses of the central nervous system and thereby increases their content in the synaptic cleft. Clinically, this effect of the drug is manifested in improved mood, a sensation of a surge of strength, increased performance. Tetanus toxin blocks the release of inhibitory neurotransmitters (GABA, glycine) in the central nervous system and thereby sharply reduces their content in the synaptic cleft, which is clinically manifested by the development of seizures;
Block or stimulate reuptake of neurotransmitters by the presynaptic membrane and, therefore, increase or decrease the concentration of neurotransmitters in the synaptic cleft.
For example, the tricyclic antidepressant imipramine blocks the reuptake of the neurotransmitter norepinephrine by the presynaptic membrane and thereby sharply increases its concentration in the synaptic cleft. Clinically, this effect of imipramine is manifested by improved mood, increased mental and physical activity;
Stimulate or block the activity of enzymes that destroy the neurotransmitter in the synaptic cleft.
For example, the anticholinesterase drug physostigmine reduces the activity of the enzyme acetylcholinesterase, which destroys the neurotransmitter acetylcholine in the synaptic cleft, and thereby contributes to an increase in its concentration, which can be clinically manifested, in particular, by a decrease in intraocular pressure and constriction of the pupil.
Stimulate or block postsynaptic receptors, i.e. imitate or block the effect of neurotransmitters.
For example, narcotic analgesics that excite postsynaptic opioid receptors and thereby mimic the affect of neurotransmitters - enkephalins. Strychnine, by blocking the receptors of the inhibitory neurotransmitter glycine, impedes the realization of its inhibitory effect; as a result, strychnine in high doses causes seizures.

Fig. 1.6. Schematic representation of the localization of pre- and postsynaptic receptors as exemplified by the adrenergic synapse (explanation in
NM - neurotransmitter; M 2 (-) - cholinergic "inhibitory" presynaptic heteroreceptor; β 1 (+) - adrenergic “activating” presynaptic autoreceptor; β - adrenergic postsynaptic receptor
In addition to receptors located on the postsynaptic membrane, i.e. postsynaptic receptors, receptors located on the presynaptic membrane, i.e. presynatic receptors (Fig. 1.6). Despite the fact that both pre- and postsynaptic receptors can be excited by the same neurotransmitter, the functional role of these receptor formations in synapses is different. If postsynaptic receptors are the final link for the transmission of a nerve impulse to an effector organ, i.e. provide unidirectional conduction of a nerve impulse from the center to the periphery, then presynaptic receptors take part in
regulation of the neurotransmitter activity of the synapse, i.e. to some extent affect the processes of release and / or synthesis of a neurotransmitter in it. It must be emphasized that presynaptic receptors do not directly participate in the conduction of a nerve impulse from a neuron to an effector organ.
Presynaptic receptors are divided into two large groups: auto- and heteroneuromodulatory receptors (see Fig. 1.6).
Presynaptic autoreceptors include receptors that are excited by their own neurotransmitter for this synapse.
For example, in synapses localized in the area of \u200b\u200bcontact between the somatic nerves and the striated muscle, when there is an excess of acetylcholine in the synaptic cleft, interacting with presynaptic autoreceptors, it inhibits the release of a new portion of the neurotransmitter excitation of presynaptic autoreceptors regulates the release of acetylcholine from the presynaptic terminals.
However, on the presynaptic membrane, in addition to autoreceptors, i.e. receptors that are sensitive to a neurotransmitter that transmits excitation in a given synapse, receptors that are not sensitive to a neurotransmitter that transmits excitation in a given synapse, but interact with another type of neurotransmitter, can be located.
For example, on the synaptic presynaptic membrane, in which acetylcholine is the neurotransmitter, presynaptic receptors sensitive to the neurotransmitter norepinephrine can be located. This type of presynaptic receptor is called heteroneuromodulating receptor.
Thus, the synapse is a complex anatomical and functional formation that ensures the transmission of a nerve impulse from a neuron to a neuron or from a neuron to an effector cell.
The sequence of the functional activity of the synapse (stages of synaptic transmission) is as follows:
Synthesis and accumulation of a neurotransmitter in vesicles localized in presynaptic thickenings (synthesis of a neurotransmitter occurs not only in presynaptic thickenings, but also in a neuron and axons);
The release of the neurotransmitter into the synaptic cleft at the time of passage of a nerve impulse;
The interaction of the neurotransmitter with postsynaptic receptors, which entails the activation of receptors and a change in the functional activity of the effector cell;
Inactivation of the neurotransmitter (enzymatic) and / or its reuptake by the presynaptic membrane, i.e. restoration of the ability of the synapse to again transmit a nerve impulse to an effector cell.
Synapses have the following basic properties:
Unilateral conduction of excitation (a nerve impulse can pass only from the presynaptic membrane to the postsynaptic one);
Synaptic delay, i.e. a certain time is spent on the transmission of a nerve impulse in the synapse. (The speed of synaptic transmission is on average more than 10 times lower than the speed of propagation of a nerve impulse through the nerve. For a chemical synapse, it usually ranges from 0.2 -0.5 ms);
Fatigue - a gradual decrease or complete cessation of transmission of a nerve impulse with prolonged nervous stimulation. The basis of this phenomenon is, on the one hand, the depletion of neurotransmitter reserves in presynaptic thickenings, and on the other hand, a decrease in sensitivity to postsynaptic receptors to the neurotransmitter;
High sensitivity of synaptic formations to drugs and poisons.
It is on the last property of synapses that the entire pharmacology of drugs that influence the functional activity of synapses located in various organs and tissues of the body is based. It must be emphasized that the object of pharmacological action can be any of the stages of synaptic transmission. As drugs that affect synaptic transmission, exogenous analogues of neurotransmitters, their chemical precursors, and other biologically active substances that can in any way alter the functional activity of the synapse are used.
It should be noted that many drugs have not one but several points of application of the effect at the synapse level. So, for example, the antidepressant pirlindol not only inhibits the activity of the monoamine oxidase enzyme in the synaptic cleft, but also blocks the reuptake of norepinephrine by the presynaptic membrane.
In relation to the localization of the receptor to the synapse, they can be divided into presynaptic, postsynaptic and extrasynaptic. The latter, for example, include receptors located on the cell membranes of platelets.
From the point of view of cell topography (location), receptors can also be classified according to their location on cellular structures as follows:
membrane receptors - receptors located on the cytoplasmic membrane;
cytosolic receptors - receptors located on intracellular formations;
nuclear receptors - receptors located on the membrane of the cell nucleus.
As noted earlier, as a result of interaction with the receptor of endogenous biological substances or drugs, the functional activity of target cells changes. This process can be implemented in different ways, strictly defined for different types of receptors. In accordance with this, four types of receptors are currently distinguished, each of which has its own, fundamentally different from the others, mechanism by which a signal from the receptor initiates a cascade of biochemical and / or biophysical reactions leading to a change in the functional state of target cells.
The first three types of receptors are localized on the cell (cytoplasmic) membrane, and the fourth type of receptors includes cytosolic and nuclear receptors.
Type I receptors include cellular (membrane) receptors that realize their effects through the so-called signaling G-proteins (Fig. 1.7).
At the first stage, a biologically active substance or drug, "going up" to the cell membrane, "recognizes" the receptor and interacts with it, after which the receptor activates a specialized signal G protein located on the inner surface of the membrane. Further, the activated G-protein changes the functional activity of the internal effector element, which, as a rule, is enzymes. Then the effector element, which is an enzyme, activates a secondary messenger, or a secondary messenger, which triggers a cascade of biochemical reactions that change the functional activity of target cells.
Type I cell receptors, i.e. receptors conjugated with signal G-proteins are structurally similar to each other, and in their spatial organization they are serpentine (from French serpantine - snake, ball) structure (Fig. 1.8).
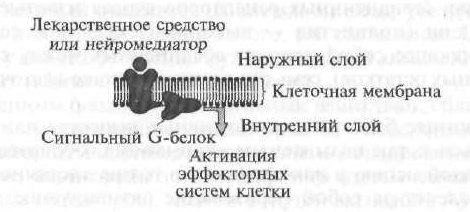
Fig. 1.7. The structure of the receptor type 1 (explanation in the text)
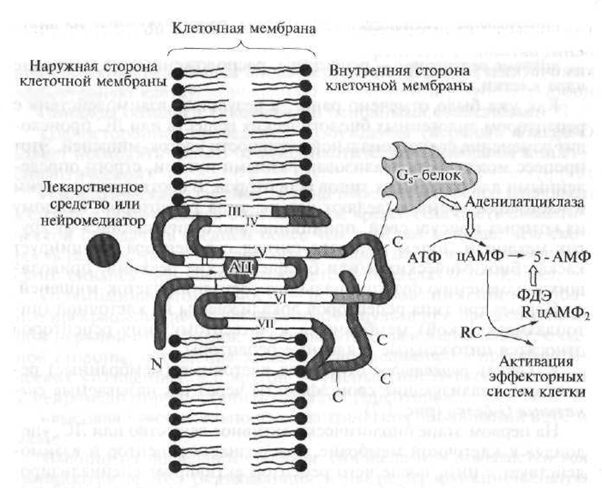
Fig. 1.8. Schematic representation of the structure of the "serpentine"
receptor:
N is the polypeptide part of the receptor located above the cell membrane; C is the polypeptide part of the receptor located under the cell membrane; AC - the active center of the receptor with which the drug interacts; go neurotransmitter; ATP - adenosine triphosphate - secondary messenger; cAMP - cyclic adenosine monophosphate; 5-AMP - adenosine-5 "monophosphate; PDE - phosphodiesterase; R. RC - cAMP-dependent enzyme (protein kinase) with regulatory and catalytic (accelerating the reaction) subunits; 1-VII - polypeptide chains of the serpentine receptor
Serpentine receptors include convoluted polypeptide chains (a polypeptide is a high molecular weight compound, which is a chain of interconnected amino acid residues) that penetrate the cell membrane seven times.
Endogenous biologically active substances or drugs can bind to the so-called "pocket" formed by the polypeptide chain and located in the thickness of the cell membrane, which entails the formation of an activating signal, which is transmitted to parts of the receptor chain located in the cytoplasm of the cell. Signal G proteins interact with cy-
stol (intracellular) sections of the floor and peptide chain; activate and launch a cascade of biochemical reactions that change its functional activity about the target cell, i.e. initiate a primary pharmacological response.
Currently, several types of signal G proteins are known.
Signal G, -proteins. These signaling proteins, as a rule, activate an effector element - the enzyme adenylate cyclase, which in turn stimulates the synthesis of a second messenger, cyclic adenosine monophosphate (cAMP) in the cell (from ATP). The biological role of cAMP as a secondary messenger is very important. For example, an increase in its content in heart cells entails an increase in the frequency and strength of heart contractions. In addition, an increase in the concentration of cAMP in various target cells causes relaxation of the smooth muscles of the vessels and bronchi, mobilization of energy reserves (decay of carbohydrates in the liver), inhibits the aggregation ability of platelets, lowers the tone of the myometrium (uterine muscle) and bladder, etc.
A number of neurotransmitters, such as adrenaline (by activating β-adrenoreceptors), dopamine (by activating D 1 -dopamine receptors), adenosine (by activating adenosine A 2 receptors), belong to endogenous biologically active substances with the ability to activate signaling G s-proteins, histamine (by activation of histamine G 2 -receptorop), serotonin (by activation of serotonin 5-HT 4 -receptors), as well as a number of hormones, for example, vasopressin (by stimulation of V 2 -vasopressin receptors), etc.
Signal G i -proteins. Unlike signal G s proteins, the activation of signal G i proteins does not stimulate, but inhibits, the activity of the effector element, the adenylate cyclase enzyme, which entails a decrease in the concentration of cAMP in the target cells of the secondary messenger. A decrease in the content of cAMP in target cells causes a decrease in cardiac contractions, an increase in the tone of blood vessels and bronchi, i.e. the opposite effect to the increase in cAMP content on target cells. In addition, a number of signaling G i proteins is involved in the regulation of the functional activity of transmembrane ionic Ca 2+ and K + channels.
A number of neurotransmitters, for example, adrenaline and norepinephrine (by activating a 2 -adrenoreceptors), dopamine (by activating D 2 - dopamine receptors), adenosine (by activating A 1, belong to endogenous biologically active substances that are capable of activating signal G i-proteins. adenosine receptors), acetylcholine (by activation of M 2 and M 4 muscarinic receptors), etc.
Signal G ^ proteins. These signaling proteins contribute to the activation of another effector element of the target cells, the phosphorylase C enzyme, which in turn stimulates the formation of secondary messengers, diacylglycerol (DAG) and inositol-1,4,5-triphosphate (ITP) in the target cells. The first of them (DAG) is associated with the cell membrane and initiates biochemical reactions involved in the regulation of contractile status, cell growth and division, and secretion of certain hormones by target cells. Under the influence of the phospholipase A 2 enzyme, DAG can be metabolized to arachidonic acid, which takes part in the synthesis of biologically active substances such as eicosanoids - prostaglandini, prostacyclins, thromboxanes, leukotrienes (see T. I, p. 478).
The second secondary messenger, ITF, is not fixed on the cell membrane and moves into the intracellular medium (cytosol), where it initiates the release of Ca 2+ ions from cell depots, i.e. promotes the transition of inactive Ca 2+ ions to the active form.
Many researchers consider Ca 2+ ions as a tertiary messenger, or intermediary. This is due to the fact that the role of Ca 2+ ions in the regulation of the functional activity of cells is very important. Ca 2+ ions can enter the cell from the external environment through special transmembrane ion channels and / or released from cell depots. The main depot (the site of accumulation of inactive Ca 2+ ions) in the cell is the endoplasmic, or sarcoplasmic, reticulum (reticulum sarcoplasmaticunr, synonym: endoplasmic reticulum - intracellular organelle, which is a system of tubules and cisterns located in the cytoplasm, limited by the membrane; takes part in providing transport of substances in the cytoplasm). Free (active) Ca 2+ ions coming from the sarcoplasmic reticulum to the cytoplasm interact with some Ca 2+ binding proteins, the most important of which is calmodulin. The complex "calmodulin-Ca 2+" and / or complexes of Ca 2+ ions with other calcium-binding proteins trigger a cascade of biochemical reactions in the cell. As a result, depending on the target organs in which this process occurs, an increase in the contractile function of the myocardium and skeletal muscle, an increase in the tone of the smooth muscles of blood vessels, bronchi, and uterus, an increase in the secretory activity of glandular tissue, stimulation of the release of neurotransmitters from nerve endings, etc., are initiated. . It has also been proven that Ca 2+ ions have the ability to increase the activity of enzymes involved in protein, carbohydrate and fat metabolism.
In addition to the direct relationship between secondary messengers - DAG and ITF and, therefore, signal G q proteins, Ca 2+ ions under physiological conditions have a rather complex interaction with the cAMP secondary messenger, the activity of which is regulated by signal G s and G i proteins. Thus, it was shown that free Ca 2+ ions entering the cytoplasm of a nerve cell through the calmodulin-Ca 2+ system initiate a decrease in the content of iAMP in the cell. At the same time, to maintain the open state of calcium ion channels in the cell, high cAMP concentrations are necessary, i.e. the decrease in cAMP content initiated by the calmodulin-Ca 2+ complex entails a cessation of the intake of free Ca 2+ ions in the cytoplasm. On the other hand, there is evidence that the cAMP secondary messenger enhances the absorption of free Ca 2+ ions by the sarcoplasmic reticulum, i.e. promotes the transition of Ca 2+ ions from the free, active form to the bound, inactive form.
As a result of the increase in the content of secondary messengers in the target cells - DAG and ITF - the tonus of smooth muscles increases, the secretion of glands increases, the release of neurotransmitters from presynaptic endings is facilitated, the aggregation ability of platelets, etc.
Endogenous biologically active substances with the ability to activate C q signal proteins include neurotransmitters such as norepinephrine (by activating a 1 -adrenoreceptors), acetylcholine (by activating muscarinic M 1 and M 3 receptors), serotonin (due to activation of serotonin 5-HT 2a receptors), histamine (due to activation of histamine H 1 receptors), as well as other endogenous biologically active substances, for example, bradykinin and angiotensin.
Currently, in addition to the listed signal G-proteins (G s, G |, G q), other signal G-proteins, G s, G i, G q, have been identified, the physiological role of which is still completely unclear. But at the same time, there is evidence that, for example, the signal C o protein is involved in the regulation of the functional activity of transmembrane ion channels.
The functional unit of type 11 receptors is a transmembrane (penetrating the entire thickness of the cell membrane) protein (enzyme). The receptor itself consists of two identical fragments, which are called monomers. The monomers are located at an insignificant distance from each other, and the monomer itself consists of two functionally active subunits - domains, interconnected by a polypeptide segment that intersects the lipid bilayer membrane (Fig. 1.9). The a-subunit of the monomer protrudes above the outer surface of the membrane and is responsible for binding of the receptor to biologically active substances, and the P-subunit is immersed in the cytoplasm of the cell.

Fig. 1.9. The structure of the receptor type II (explanation in the text): 1 - a-subunit of the monomer; 2 - β-subunit of the monomer
After binding of the biologically active substance to the α-subunit of the receptor, the receptor changes from an inactive monomeric state to an active dimeric state in which two monomers combine in the plane of the membrane (see Fig. 1.9). In this case, the enzymatic activity of the cytoplasmic β-subunit of the receptor is stimulated, as a result, a cascade of biochemical reactions that change its functional state is launched in the target cell.
As a transmembrane enzyme forming the receptor, as a rule, such enzymes as tyrosine kinase or guanylate cyclase are used.
An example of a tyrosine kinase receptor is insulin receptors (see T. 1, p. 435).
The guanylate cyclase signal transmission pathway begins with the interaction of the α-subunit of the receptor with an endogenous biologically active substance, for example, with atrial natriuretic factor (ANF), which is a biologically active substance secreted by atrial cells and involved in the regulation of cardiac contractions. As a result of this interaction, a change in the configuration of the receptor occurs, which consists in combining its monomers into a dimer. This process activates the enzyme part of the receptor located in its cytosolic β-subunit, i.e. the guanylate cyclase enzyme, which in turn promotes an increase in the concentration of the cyclic guanidine-3,5 "monophosphate (cGMP) secondary messenger in the target cell. An increase in the concentration of cGMP in target cells triggers a cascade of biochemical reactions that change their functional state, for example, relaxation of smooth muscle cells vessels.
Type III receptors include receptors that, under the influence of endogenous biologically active substances - neurotransmitters, ensure the passage of the corresponding ions through the cell membrane, which leads to a change in its (membrane) electric charge (potential).
In their structure, III tina receptors represent a channel that penetrates the lipid bilayer of the cell membrane, formed by several poly-piped units (Fig. 1.10). For example, the nicotinic (H) receptor is a channel with a diameter of 8 nm, formed by five polypeptide subunits (a - two, β, γ, d) (see Fig. 1.10). When the neurotransmitter acetylcholine interacts with a portion (domain) protruding above the surface of the cell membrane, the α-subunit of the receptor, its structure changes and a central channel opens, through which Na + ions enter the target cell according to the concentration gradient, which entails a change in its functional activity. In addition to H-cholinergic receptors, receptors for gamma-aminobutyric acid and excitatory amino acids belong to type III receptors.
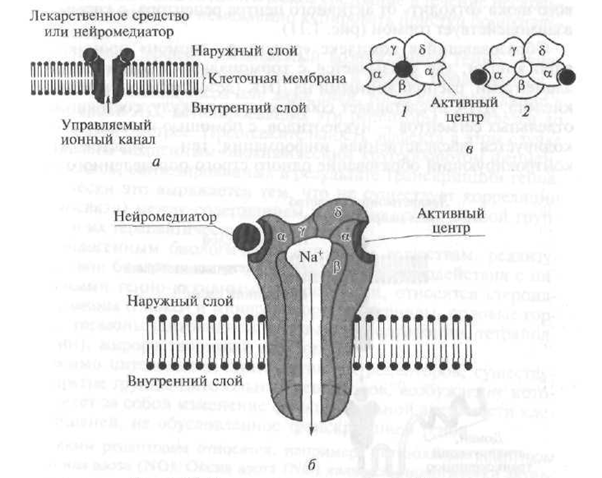
Fig. 1.10. The structure diagram of the receptor type 111:
a is a circuit diagram; b - transmembrane ion channel (in the context); c - transmembrane ion channel (top view); / - channel in inactive (closed) state; 2 - channel in the active (open) state; a. β, γ, d - channel polypeptide subunits
Type IV receptors include intracellular and nuclear receptors. The biologically active substances interacting with this type of receptor are lipophilic (readily soluble in fats) compounds, therefore, they easily penetrate the cell membrane and reach their intracellular receptors. Intracellular receptors include receptors for hormones, as well as other biologically active substances.
The mechanism of interaction of hormones with intracellular receptors is quite complicated, however, it can be schematically represented as follows. By structure, the intracellular receptor for hormones is a polypeptide consisting of several functional units - domains. In the absence of hormone, the receptor is inactive due to the fact that its active center is blocked by a specialized protein - the so-called heat shock protein. In the case when the hormone “approaches” the receptor, the heat shock protein “departs” from the active center of the receptor, with which the hormone interacts (Fig. 1.11).
The resulting receptor-hormone complex penetrates the nucleus of the cell, where it binds to hormone-sensitive elements located on DNA (deoxyribonucleic acid; DNA is a macromolecule consisting of individual segments - nucleotides, with the help of which hereditary information is encoded in the genes; the gene - a piece of DNA that controls the formation of one strictly defined protein
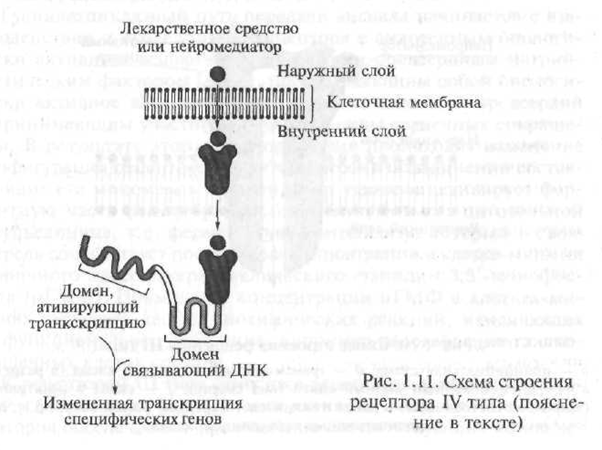
Fig. 1.11. Scheme of the structure of receptor type IV (explanation in the text)
ka). As a result of this interaction, the process of gene transcription is started - the process of transferring the information contained in the genetic code from the DNA molecule to the molecule of information RNA (mRNA, syn: matrix RNA - mRNA). Transcription is the first step in the formation of proteins in a cell. The resulting mRNA. leaves the cell nucleus and moves to the ribosomes - intracellular organelles responsible for the synthesis of protein in the cell. In the special medical literature, receptors, the activation of which causes the process of gene transcription, are referred to as genetically active receptors.
As a rule, the response of target cells to the excitation of gene-active receptors develops rather slowly, which is of very important clinical importance.
First, the response of target cells is delayed in time, since it requires the synthesis of new proteins, which usually takes 20-30 minutes, i.e. hormones, activating type IV receptors, are not able to change the pathological state within a few minutes, for example, immediately stop an attack of bronchial asthma.
Secondly, the effect caused by the excitation of gene-active receptors is quite long and can last for several hours or even days, while the plasma content of drugs that activate these receptors decreases to zero much faster. The duration of the effect in this case is due to the slow biochemical circulation of enzymes and proteins synthesized as a result of gene transcription. Clinically, this is expressed by the fact that there is no correlation (relationship) between the plasma content of a given group of drugs and their therapeutic effect.
Endogenous biologically active substances that realize their biological effects by interacting with cytosolic gene-active receptors include steroid hormones (gluco- and mineralocorticosteroids, sex hormones), thyroid hormones (triiodothyronine, tetraiodothyronine), and fat-soluble vitamin D.
In addition to cytosolic gene-active receptors, there are other groups of cytosolic receptors, the excitation of which entails a change in the functional activity of target cells, not due to transcription of genes.
Such receptors include, for example, cytosolic receptors for nitric oxide (N0). Nitric oxide (N0) is a biologically active substance formed in the vascular endothelium. As an endogenous biologically active substance, nitric oxide was first isolated from rabbit ports by the American physiologist R.F. Furchgott in 1987 and was called "endothelial relaxing factor - ORF." Nitric oxide is a lipophilic compound that easily penetrates the cell membrane, where it interacts with its specific cytosolic receptors, which entails the activation of the guanylate cyclase enzyme. The latter, in turn, stimulates the synthesis of the cGMP secondary messenger, which triggers a cascade of intracellular biochemical reactions leading to relaxation of target cells, vascular smooth muscle cells.
Thus, at present, four principal mechanisms and, accordingly, IV receptor types are distinguished, due to the interaction with which endogenous biologically active substances and / or their synthetic analogues, i.e. Drugs can affect the functional state of target cells.
However, this does not mean that the number of known receptors for biologically active substances is limited to 4. They are immeasurably greater. This is due to the fact that, through the same fundamental mechanism of action, a very large number of endogenous biological substances of various chemical structures can influence the functional activity of cells. For example, the neurotransmitters norepinephrine and histamine, which are different in their chemical structure and, therefore, in the receptors with which they interact, transmit an excitatory signal to target cells by the same basic mechanism - stimulation of the activity of signal G proteins, i.e. both interact with type I receptors.
Therefore, all currently known receptors are classified based not only on the characteristics of signal transmission to the intracellular structures of target cells, but also on the names of those endogenous biologically active substances with which they specifically interact.
It should be noted that the receptors got their name taking into account the names of endogenous biologically active substances with which they interact, long before the mechanisms of signal transmission to target cells became known.
Endogenous biologically active substances that realize their effects through interaction with their specific receptors include neurotransmitters (acetylcholine, norepinephrine, dopamine, histamine, serotonin, etc.), hormones, biologically active substances of tissue origin - autocaids (prostaglandins, thromboxanes, leukotrienes, bradykinin, angiotensin, etc.). In the special medical literature, all these substances are often combined under the term “ligands” (from Lat. Ligo - to bind, that is, a substance capable of binding to the receptor).
Thus, receptors get their name from the name of their specific ligands. For example, receptors for the dopamine neurotransmitter are called dopamine, insulin hormone - insulin, leukotriene augocaid - leukotriene, etc.
In the future, in the text of the textbook, in order to avoid confusion in terms when it comes to the mechanism of signal transmission from the receptor to intracellular formations, the term “receptor type” will be used, and when it comes to the name of the receptor due to the ligand interacting with it , the term “receptor species” will be used.
As a rule, many receptors of the same species are divided into several subtypes, for example, adrenergic receptors are divided into a- and β-adrenergic receptors, cholinergic receptors into M- and N-cholinergic receptors, etc. In most cases, subspecies are also subdivided into smaller groups: β 1 - and β 2 -adrenoreceptors, N n - and N m -cholinergic receptors, etc.
The identification of subspecies of receptors and the study of the mechanisms by which endogenous biologically active substances interact with them is very important for modern pharmacology, as it allows the creation of drugs that interact with a strictly defined subspecies of the receptor. So, for example, the division of β-adrenergic receptors into β 1 (mainly localized on the cell membrane of heart cells) and β 2 (localized, for example, on the cell membranes of smooth muscle cells of the bronchi), made it possible to create drugs that selectively affect the heart muscle (β 1 -adrenostimulants ) - the drug nonachlazine, and selectively affecting the smooth muscles of the bronchi (β 2 -adrenostimulants) - the drug salbutamol, etc.
It should be noted that the organs and tissues of the body do not contain a constant number of receptors and / or their subspecies, i.e. it is variable. Both pathological processes and drugs can change the number of receptors in an organ.
For example, coronary heart disease is accompanied by an increase in the number of 3-adrenergic receptors in the heart muscle, and in patients with hypertension, the number of both a- and β-adrenergic receptors increases. The antidepressant imipramine, with prolonged use, reduces the amount of β-adrenergic receptors in the brain tissue. There are quite a few such examples.
The affinity of endogenous (produced in the body) neurotransmitters or drugs for receptors is characterized by the term “affinity”, and the speed and strength of their binding to receptors is designated by the term “affinity”.
Naturally, the interaction of drugs with the receptor is not an end in itself, but should lead to some change in the activity of organs or tissues of the body.
Such a change, or reaction, corresponding to the functional significance of this receptor, is called the internal activity of drugs.
Medicines with intrinsic activity and receptor affinity are agonists, i.e. act like endogenous biologically active substances.
For example, phenylephrine, a stimulator of a-adrenergic receptors, has an effect on arterioles similar to the neurotransmitter norepinephrine, i.e. he is an agonist of a-adrenergic receptors. In the special medical literature, in addition to the term “agonist”, the term “receptor stimulant” or “mimetic” is sometimes used, for example, an adrenomimetic, i.e. Drugs that stimulate adrenergic receptors.
Drugs that have receptor affinity, but inhibit exogenous and endogenous agonists from interacting with the receptor, are called antagonists.
For example, the atropine M-cholinergic receptor blocker interferes with the acetylcholine neurotransmitter M-cholinergic receptor interaction; blocker (propranolol J-adrenergic receptors, blocking β 2 -adrenostructures of the lungs, inhibits the stimulating effect of β 2 -adrenostimulator of salbutamol on them, i.e. atropine and propranolol are antagonists of the corresponding receptors.
In the specialized medical literature, in addition to the term “antagonist”, the term “receptor blockers” or “lytics”, for example, anticholinergics, i.e. drugs that block cholinergic receptors.
Schematically, the interaction of receptors with agonists and antagonists is shown in Fig. 1.12.
Agonists can exert both direct and indirect, i.e. indirect action.

Fig. 1.12. Scheme of interaction of the receptor with the agonist (a) and the antagonist (6) with the receptor (explanation in the text)
For example, an opioid receptor agonist - the drug morphine - realizes its affects by directly stimulating (mu) p-, (kappa) k- and (delta) 8-opioid receptors, i.e. exerts a direct stimulating effect on them, while the sympathomimetic ephedrine realizes its cellular effects by indirectly or indirectly stimulating a- and β-adrenergic receptors: it helps displace the neurotransmitter norepinephrine from presynaptic nerve endings, inhibits its reuptake by nerve endings, and increases the sensitivity of a- and p- adrenoreceptors to norepinephrine and epinephrine and stimulates the release of adrenaline from the adrenal cortex. Thus, ephedrine realizes its pharmacological effects not directly, but through a neurotransmitter, while it does not directly interact with the receptor.
Antagonists, as well as receptor agonists, can realize their pharmacological effects directly or indirectly, blocking the corresponding receptors.
For example, the gastric H 3 histamine receptor blocker, ranitidine, interacts directly with the gastric H 2 histamine receptors, inhibits the basal secretion of hydrochloric acid, inhibiting the interaction with the endogenous histamine neurotransmitter receptor, i.e. directly blocks the receptor, while the sympatholytic reserpine, entering the body, like the sympathomimistic ephedrine “ejects” the neurotransmitter norepinephrine from presynaptic endings, while simultaneously blocking the synthesis of new “portions” of norepinephrine, which leads to a rapid depletion of mediator reserves in presynaptic endings. As a result, a state is created when the a-adrenergic receptors of the vessels "do not work" due to the lack of their physiological stimulator noradrenapine. We can say that the sympatholytic reserpine causes a functional, indirect blockade of a-adrenergic receptors.
Some drugs combine the properties of agonists and antagonists, i.e. under certain conditions, the same receptors excite or block. In those cases when the stimulating component prevails in the pharmacological effect of the drug, they say that the drug is a partial or partial agonist.
The mechanism of action of partial or partial agonists is that these drugs, approaching the receptor, are fixed on it. However, due to the peculiarities of their chemical structure, they excite it in such a way that they cause not a complete, but only a partial stimulating response. At the same time, they interfere with the interaction of other drugs and / or the corresponding neurotransmitter with this receptor.
For example, nalorphine, which is used to treat an overdose of narcotic analgesics, is a partial agonist. Nalorfin, "going up" to the opioid receptor, displaces morphine from its connection, that is, it stops the agonistic effect of morphine on the receptor. Contacting the receptor, nalorphine to a small extent has a stimulating effect on it, which is not able to suppress the activity of the respiratory center, i.e. has a partial agonistic effect on the opioid receptor, in parallel with this, it blocks it for morphine, thereby exhibiting an antagonistic effect with respect to morphine. The disadvantage of nalorphine is that in case of an overdose, its stimulating effect on the receptor is enhanced and it inhibits the respiratory center, as well as morphine.
In those cases when a blocking effect prevails in the pharmacological effect of drugs, they say that the drug is an antagonist with its own or internal activity.
Peculiarities of the mechanism of action of antagonists with their own activity can be considered on the example of the β 1 -adrenoblocker of acebutalol. Acebutalol belongs to the group of β 1 -adrenergic blockers with their own internal sympathomimetic activity, i.e. combines the properties of a blocker (lytic) and a stimulant (mimetic) of β 1 -adrenoreceptors.
The mechanism of action of acebutalol is due to the peculiarities of its chemical structure, due to which the drug, on the one hand, blocks the myocardial β 1 -adrenoreceptors, and on the other hand, stimulates their activity, thereby simulating the physiological effect of mediators - catecholamines (see T. 1. p. 170).
All drugs according to the peculiarities of their action can be divided into two large groups - with specific and non-specific effects.
Drugs that have a non-specific effect include drugs that have a wide range of pharmacological activity and various points of application of effects. This group includes vitamins, biogenic stimulants, antioxidants, adaptogens, etc.
Drugs with a specific effect include drugs characterized by the properties of agonists or antagonists of the corresponding receptors, i.e. the basis of their mechanism of action is the ability to interact with specific receptors for them.
However, it should be noted that the pharmacological effect of drugs that have a specific effect on any receptors will not always be highly specialized, for example, reducing heart rate (HR) or suppressing secretion of gastric juice. The total pharmacological effect of many drugs will depend on how many and in which organs and tissues of the body specific receptor formations for this drug are localized.
For example, β-adrenergic receptors are located in the heart muscle, vascular wall, bronchi, uterus, adipose tissue, skeletal muscle, etc. As a result of this, drugs that stimulate β-adrenergic receptors will, to a certain extent, cause an increase in the strength and frequency of heart contractions, expansion of blood vessels, bronchi, and lower tone of the uterus, i.e., have a systemic effect on the body.
The specific effect of drugs can be selective or selective and, accordingly, non-selective or non-selective. The selectivity of drugs is determined by whether it affects all subspecies of any receptors or their specific subspecies.
For example, the non-selective β-adrenergic blocker propranolol blocks both β 1 -adrenoreceptors located in the myocardial tissue and β 2 -adrenoreceptors located, in particular, in the lung tissue, i.e., has a non-selective (non-selective) effect on both subspecies of β-adrenergic receptors . whereas the selective β 1 -adrenergic blocker atenolol in therapeutic doses blocks only β 1 myocardial adrenergic receptors, i.e. exerts a selective (selective) effect on a specific β-adrenergic receptor.
Clarification of the physiological role and localization of receptor subspecies allows the creation of highly effective drugs that selectively act on different subspecies of receptors.
For example, drugs that block β 1 -adrenergic receptors are widely used in the clinic for the treatment of coronary heart disease, arterial hypertension, and drugs that stimulate β 2 -adrenergic receptors have found their application for the treatment of bronchial asthma. Medicines that block H 1 histamine receptors are preferably prescribed in clinical practice to prevent and / or arrest allergic reactionswhile drugs blocking H 2 histamine receptors are effective in treating gastric and duodenal ulcers.
The creation of drugs with a selective effect, has largely optimized their clinical use, as they significantly reduced side effect.
The basis of the mechanism of action of drugs, as a rule, is their ability to initiate (trigger) complex biochemical and / or biophysical processes that ultimately alter and / or optimize the functional activity of the target cell. Medicines can realize their effect on organs and / or target cells by: direct chemical interaction; physico-chemical interaction on the cell membrane; actions on specialized enzymes; actions on regulatory genes; actions on specific receptors.
Direct chemical interaction of drugs.
This mechanism of action of drugs is quite rare and can be realized outside the cell, for example, in the lumen of the stomach or intestines. Its essence lies in the fact that drugs enter into a direct chemical reaction with molecules and / or ions that are formed in the body in a normal or pathological condition. An example of direct chemical interaction is the chemical reaction of neutralization of hydrochloric acid of the stomach when taking antacid drugs.
Physico-chemical interaction of drugs on the cell membrane.
One of the main functions of the cytoplasmic membrane is the implementation of ion exchange between the cytoplasm and the extracellular environment. Transmembrane ion exchange can also take place through special voltage-dependent transmembrane ion channels - sodium, potassium, calcium, chlorine, etc. Some drugs, reaching the cell membrane, interact with these channels and alter their functional activity. So, for example, the antiarrhythmic action of class IA drug - quinidine - is based on its ability to block the passage of Na + ions through transmembrane sodium channels.
The effect of drugs on specialized enzymes.
A relatively small amount of drugs realizes its pharmacological effect by changing the activity of some specialized cellular enzymes. Medicines that increase the activity of cellular enzymes are called inductorsenzymes. Such action is possessed, for example, by sleeping pills and anticonvulsant drug phenobarbital, which significantly enhances the activity of microsomal liver enzymes. The biological significance of this effect of phenobarbital and related drugs will be discussed below.
Medicines that inhibit the activity of specialized enzymes are called inhibitorsenzymes. So, for example, an antidepressant from the group of monoamine oxidase inhibitors (MAO), the drug pirlindole realizes its antidepressant effect by inhibiting the activity of the MAO enzyme in the central nervous system. The ability to inhibit the activity of the enzyme acetylcholinesterase is the basis of the pharmacological activity of anticholinesterase drugs, for example physostigmine. It is known that under physiological conditions, acetylcholinesterase inactivates (destroys) acetylcholine, a neurotransmitter that transmits excitation in the synapses of the parasympathetic nervous system. Physostigmine, suppressing the activity of acetylcholinesterase, promotes the accumulation in the synapses of the parasympathetic system of the neurotransmitter acetylcholine, as a result of which the tone of the parasympathetic nervous system increases, which is manifested at the systemic level by the development of bradycardia, lowering blood pressure (BP), and increasing motility of the gastrointestinal tract (GIT), pupil, etc. Medicines can interact reversibly and irreversibly with enzymes. For example, the drug enalapril reversibly inhibits the activity of the angiotensin-converting enzyme, which entails, in particular, a decrease in blood pressure, while organophosphorus toxic substances irreversibly inhibit the activity of acetylcholinesterase. The effect of drugs on regulatory genes.Currently, scientists are making attempts to create drugs that realize their pharmacological effects by directly affecting the physiological activity of regulatory genes. This trend seems especially promising after the structure of the human genome was deciphered in 2000. It is believed that the selective normalization of the function of regulatory genes under the influence of drugs will make it possible to achieve success in the treatment of many, including previously incurable, diseases. The effect of drugs on receptors.Before moving on to the characteristics of the interaction of drugs with receptors, it is necessary to clarify what we mean by the term “receptor” (from lat. recipio - take, take). From the course of physiology it is known that the term "receptor" means highly specialized formations that are able to perceive, transform and transmit the energy of an external signal to the nervous system. These receptors are called sensory(from lat. sensus - feeling, sensation, perception). Sensory receptors include receptors of the organs of hearing, vision, smell, taste, touch, etc. Sensory receptors of these organs belong to the so-called exteroreceptors. If the presence of sensory organs that respond to external stimuli of irritation has been known since ancient times, then the presence of sensory receptors inside the body was questioned until the middle of the 19th century. For the first time, the Russian physiologist I.F. Zion, which showed in 1866 a drop in blood pressure due to aortic irritation in a rabbit experiment. This discovery gave rise to the search and study of receptors located inside the body, and these receptors themselves were called interreceptors. TObeginning of XX century a sufficient number of sensory interoreceptors was revealed and their important role in the regulation of the physiological functions of the body was proved. In 1905, J. Langley proved that when a drug is applied to a cell membrane, a pharmacological effect develops if it is applied only to a specific area of \u200b\u200bit. Moreover, this site makes up only a small part of the total area of \u200b\u200bthe cell surface. This observation allowed J. Langley to conclude that specialized receptor sites interacting with drugs exist on the cell membrane. However, the priority in creating the receptor theory of the action of drugs belongs to the German physiologist P. Ehrlich, who in 1906 first introduced the term “receptor” and formulated the postulate “the drug does not work if it is not fixed on the cell membrane”. According to the theory of P. Ehrlich, a drug molecule has two functionally active groups, one of which ensures its fixation on the cell surface in the region of the drug receptor, and the second functional group interacts with the receptor and triggers a complex chain of biochemical reactions that alter its (cell) physiological activity . Thus, as early as the beginning of the 20th century. it became apparent that there are at least two classes of interoreceptors: sensory receptorstransmitting information about the state of internal organs and body tissues in the central nervous system; cell receptorswhich interact with drugs that alter the functional activity of target cells.<>Immediately, it should be noted that in the future in the text of the textbook, in order to avoid confusion in terminology, receptors for drugs and biologically active substances, i.e. cellular, or cytoreceptorywill be denoted by the term “receptor”, while sensory interoreceptors will be denoted by a term characterizing their functional activity, for example, “baroreceptors”, “pain receptors”, etc. The discovery by P. Ehrlich on the cell membrane of drugs receptors served as a starting point for the development of pharmacological science, in particular pharmacodynamics, one of the main tasks of which is to study the receptor mechanisms of action of drugs. Currently, the structure of a large number of cellular receptors, the features of the interaction of certain biologically active compounds with them have been revealed, which made it possible, on the one hand, to understand the mechanism of action of known drugs, and on the other hand, it was the basis for the creation of new highly effective drugs. Naturally, it is difficult to imagine that in the course of evolution, receptors for various synthetic (chemically obtained) drugs were formed in the human body, especially since the vast majority of drugs presented on the modern pharmaceutical market have been synthesized in the last 50 years or less. It is proved that the receptor apparatus of the cell is a very ancient functional-structural formation. So, a- and b-adrenoreceptors (receptors, the interaction of which norepinephrine and adrenaline affect the functional activity of the cell) are found not only in animal cells, but also on the cell membranes of plant cells, for example, in the cells of the plant nittella, where a- and b- adrenoreceptors regulate the movement of protoplasm (cell contents). Then what are the receptors for drugs discovered by P. Ehrlich, and why do they interact with them? Currently, there is no doubt that the so-called drug receptors are actually receptors for endogenous (produced in the body) biologically active substances involved in the regulation of the functional activity of internal organs and body tissues. Such biologically active compounds include substances released from the nerve endings at the time of transmission of the nerve signal, as well as hormones, vitamins, amino acids, etc. For each endogenous biologically active substance, there are strictly specific receptors for it. So, for example, the biologically active substance produced in the body, adrenaline, can activate strictly specific a - and b-adrenergic receptors, and glucocorticosteroids - hormones of the adrenal cortex - interact only with glucocorticosteroid receptors strictly specific to them. Synthetic drugs that realize their effects by interacting with the receptor apparatus of the cell, in their chemical structure, are more or less similar to endogenous biologically active compounds that interact with similar receptors. For example, synthetic vasoconstrictor (causing vasoconstriction) drugs phenylephrine is close in its chemical structure to the endogenous biologically active substance norepinephrine, therefore, like norepinephrine, it has the ability to stimulate a-adrenoreceptors. Sometimes, due to the peculiarities of their chemical structure, drugs can interact not with the receptor itself, but with the adjacent portion of the cell membrane. Since in this case, the drug interacts not with the receptor itself, but with the adjacent portion of the cell membrane, they speak not of an exciting or blocking effect on the receptor, but allostrichesky(from Greek allos - another, different) effect, or effect. As a result, a change in both the structure of the membrane adjacent to the receptor and individual components of the receptor itself can occur, which may entail a change in the sensitivity of the receptor to a biologically active substance specific to it. In cases where the sensitivity of the receptor to a biologically active substance increases, talk about sensitization(from lat. sensus - feeling) or about sensitization(from lat. sensibilis - sensitivity) of the receptor, and in those cases when the sensitivity of the receptor decreases, talk about desensitizationreceptor. The peculiarity of the allosteric effect lies in the fact that drugs that have this kind of mechanism of action do not directly affect the transmission of a nerve impulse, but modify it in the desired direction. For example, the mechanism of action of anxiolytics (anti-anxiety drugs; synonyms: tranquilizers), in their chemical structure being derivatives of benzodiazepine, is based on the phenomenon of allosteric excitation of postsynaptic benzodiazepine receptors. The excitation of the latter, in turn, promotes the activation of inhibitory postsynaptic receptors of gamma-aminobutyric acid (GABA receptors), which is clinically manifested by the elimination of symptoms of neurotic diseases such as anxiety, anxiety, fear, etc. Receptors, interacting with which, a biologically active substance or drug in any way changes the functional state of the target cell, are called specific. In addition to specific receptors, so-called non-specificfor drugs receptors. In the specialized medical literature, these receptors are also called the "place of loss" of drugs. By contacting such receptors, drugs do not have any biological effect, but they themselves become biologically inactive. An example of this type of receptor can serve as receptors located on plasma proteins, in particular, on water-soluble proteins - albumin. The structure of the receptors is quite complex, but most of them are protein macromolecules or glycoproteins, which may also include ions, lipids, nucleic acids, etc. Receptor i.e. the protein macromolecule forming it is characterized by a specific, specific for each receptor, spatial arrangement of its chemical groups. The protein macromolecule forming the receptor can be integrated (immersed) in the lipid bilayer of the cytoplasmic membrane or localized inside the cell. The main function of a cell receptor is to “recognize” a chemical signal transmitted to it through an endogenous biologically active substance and / or drugs and transform it into a corresponding biochemical and / or biophysical cell response. It was previously believed that drugs or endogenous biologically active substances interact with receptors of the “key and lock” type, i.e. the receptor has such a structure that allows the drug to find “your” receptor, connect to it and, as it were, “turn on” and “turn it off”. However, with the development of medical science, it became obvious that this is not entirely true. At present, the molecular processes of the conversion of extracellular signals into intracellular, regulating cell function, have already been studied quite well. mechanisms that result in the effect of the interaction of endogenous biologically active substances or drugs with receptors. When interacting with the receptor of an endogenous biologically active substance and / or an active drug like it occurs conformation- spatial change in the form of a protein macromolecule, which is the trigger for various intracellular processes that determine the response of a target cell to a mediator and / or drug. For example, the activation of b 2 -adrenoreceptors of bronchial smooth muscle under the influence of a b 2 -adrenostimulator phenoterol leads to an increase in the activity of the enzyme adenylate cyclase, which contributes to the accumulation of cyclic adenosine monophosphate (cAMP) in the cell and, as a result, cell relaxation. In general biological terms, cellular receptors can be considered as strictly specialized “sensory organs” of cells, through which they perceive “information” emanating, for example, from the central nervous system and / or endocrine system. Despite the important role of the receptor apparatus, receptors occupy only an insignificant part of the cell membrane. For example, the M-cholinergic receptor apparatus of a cell occupies no more than 1/6 000 of its surface area. Studying the characteristics of the interaction of drugs with the receptor, on the one hand, allows us to understand the basis of the molecular mechanism of its action, and on the other hand, provides information on what changes should be made in the structure of drugs to enhance its ability to interact with this receptor, i.e. . allows targeted synthesis of new highly effective drugs. Under physiological conditions, different cellular receptors do not function independently, but are in constant interaction with each other, thereby regulating the specific activity of the cell. For example, activation of cardiac b-adrenergic receptors by endogenous norepinephrine causes, in particular, an increase in the number of heart contractions, and activation of M-cholinergic receptors of cardiac cells by endogenous acetylcholine, on the contrary, causes a decrease in the number of heart contractions. A great contribution to the understanding of the receptor mechanisms of action of drugs was made by the discovery of pre- and postsynaptic receptors. Sinaps(from Greek synopsis - connection) is a specialized contact zone between nerve cells or other excitable structures of the body, which ensures the transmission of incoming information and the preservation of its informational significance. The study of the structure and functional role of synapses was begun at the end of the 19th century. after the Spanish histologist S. Ramon-i-Cajal (S. Ramon at Cajal) suggested the presence of a specialized transmission system in the central nervous system. Synapses got their name in 1897, when the English physiologist Ch. Sherrington proposed this term to refer to the area of \u200b\u200bcontact between nerve cells. Currently, there are three types of synapses: 1) “electrical” synapses, in which information is transmitted by transferring an electrical signal from a pre-synaptic membrane. This type of synapse is called efaps(from Greek ephapsis - tight contact); 2) "chemical" synapses in which information is transmitted through special biologically active substances - neurotransmitters(from Greek neuron - nerve and lat. mediator - intermediary; synonyms: mediator); 3) “mixed” synapses in which information is transmitted both chemically and electrically. The pharmacological effects of the vast majority of drugs that affect the functions of synapses are realized by their effect on a particular stage of signal transmission in chemical synapses, i.e. in synapses of the second kind. As a rule, chemical synapses are classified by neurotransmitters that transmit nerve impulses in them, as follows: synapses in which acetylcholine acts as a mediator are called cholinergic; synapses in which adrenaline and norepinephrine act as a mediator are called adrenergic; synapses in which ATP and adenosine act as a mediator are called purinergic; synapses in which gamma-aminobutyric acid acts as a mediator are called GABA-ergic, etc. The structure of the synapse is currently well understood. The synapse consists of a presynaptic process of a nerve cell (axon end) and a “signal”-receiving apparatus located on the membrane of an effector (“executive”) cell.
Pharmacodynamics - pharmacological effects, mechanisms of action, localization of action, types of action of drugs.
Pharmacological effects medicinal substance - changes in the activity of organs, body systems that cause this substance (for example, increased heart contractions, lowering blood pressure, stimulating mental activity, eliminating fear and tension, etc.).
Each substance causes a number of pharmacological effects characteristic of it. In each case, only certain effects of the drug substance are used, which are defined as the main effects. The remaining (unused, undesirable) pharmacological effects are called collateral.
Mechanisms of action drugs - the ways in which substances cause pharmacological effects are very diverse. The main options for mechanisms of action include actions on:
- specific receptors;
- enzymes;
- ion channels;
- transport systems.
Most drugs act on specific receptors. These receptors are most often represented by functionally active protein molecules, the interaction with which gives rise to biochemical reactions that lead to pharmacological effects.
There are specific receptors associated with cell membranes (membrane), and intracellular receptors (cytoplasmic, nuclear).
Membrane receptors (receptors of the cytoplasmic membrane) are divided into:
- receptors directly coupled to ion channels;
- receptors directly conjugated to enzymes;
- receptors that interact with G-proteins.
TO receptors directly coupled to ion channels, include, in particular, Ν-cholinergic receptors and GABA A-receptors.
When Ν-cholinergic receptors are stimulated (nicotine-sensitive cholinergic receptors), the sodium channels directly coupled to them open. Stimulation of Ν-cholinergic receptors leads to the discovery of Na + channels, the entry of Na + ions into the cell, depolarization of the cell membrane and an exciting effect.
GABA A receptors are directly coupled to chlorine channels. Stimulation of GABA A receptors leads to the discovery of Cl - channels, the entry of Cl - ions, hyperpolarization of the cell membrane and inhibitory effect.
TO receptors that are directly linked to enzymesinclude, in particular, insulin receptors directly conjugated to tyrosine kinase.
G-protein receptors - M-cholinergic receptors (muscarinic-sensitive cholinergic receptors), adrenergic receptors, dopamine receptors, opioid receptors, etc.
G-proteins, i.e. GTP-binding proteins, are localized in the cell membrane and consist of α-, β- and γ-subunits. When a drug interacts with a receptor, the α-subunit of a G-protein binds to GTP (GTP) and acts on enzymes or ion channels.
One receptor interacts with several G-proteins, and each complex of the α-subunit of G-protein with GTP acts on several enzyme molecules or on several ion channels. Thus, the mechanism of an amplifier (amplifier) \u200b\u200bis implemented: when one receptor is activated, the activity of many enzyme molecules or many ion channels changes.
One of the first to be found was G-proteins associated with β 1 - cardiac adrenergic receptors. When the sympathetic innervation of the heart is activated, β 1 -adrenoreceptors are excited; adenylate cyclase is activated via G-proteins; cAMP is formed from ATP, protein kinase is activated, under the action of which Ca 2+ channels are phosphorylated and opened.
An increase in the entry of Ca 2+ ions into the cells of the sinoatrial node accelerates the fourth phase of the action potential, the frequency of the generated impulses increases - heart contractions become more frequent.
The discovery of Ca 2+ channels in the fibers of the working myocardium leads to an increase in the concentration of Ca 2+ in the cytoplasm (Ca 2+ input promotes the release of Ca 2+ from the sarcoplasmic reticulum). Ca 2+ ions bind to troponin C (a component of troponin-tropomyosin); thus, the inhibitory effect of troponin-tropomyosin on the interaction of actin and myosin decreases - heart contractions intensify (Fig. 10).
Fig. 10. The mechanism of increasing and strengthening heart contractions during stimulation of β 1 -adrenoreceptors. AC - adenylate cyclase; PC - protein kinase; CA - sinoatrial node; Ttm - troponin-tropomyosin.
When parasympathetic innervation of the heart (vagus nerves) is activated, M 2 -cholinergic receptors are excited, and through G-proteins, adenylate cyclase is inhibited - heart contractions are weakened and weakened (mainly atrial contractions are weakened, since the parasympathetic innervation of the ventricles is relatively poor).
Thus, G-proteins can have a stimulating and inhibitory effect on adenylate cyclase. Stimulating G-proteins were designated as G s-proteins (stimulate), and inhibitory G-proteins (inhibit) (Fig. 11).
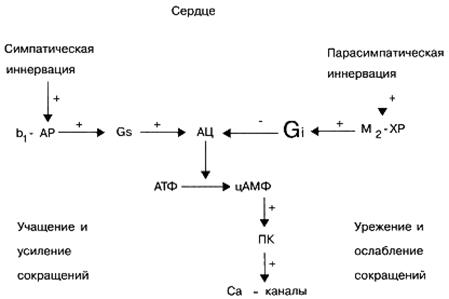
Fig. 11. The mechanism of changes in the frequency and strength of heart contractions during stimulation of the sympathetic and parasympathetic innervation.
Cholera toxin activates C s proteins (this leads to activation of adenylate cyclase and in cholera is manifested by secretion of fluid through the intestinal epithelium).
Pertussis toxin activates G i proteins.
When M 1 -cholinergic receptors, M 3 -cholinergic receptors, α 1 -adrenoreceptors are excited through G q proteins, phospholipase C is activated, which contributes to the formation of inositol-1,4,5-triphosphate and diacylglycerol from phosphatidylinositol-4,5-diphosphate .
Inositol-1,4,5-triphosphate acts on receptors of the sarcoplasmic reticulum membrane sensitive to it and stimulates the release of Ca 2+ ions from the sarcoplasmic reticulum (Fig. 12). With the stimulation of αι-adrenoreceptors of blood vessels, this leads to a contraction of the smooth muscles of the vessels and narrowing of the vessels (Fig. 13).

Fig. 12. The effect of phospholipase C on the level of cytoplasmic C a2 +.
The sensitivity of receptors to agonists and the number of receptors are constantly changing. So, after stimulation of β 1 -adrenoreceptors by a β 1 \u200b\u200b-adrenoreceptor agonist, phosphorylate by a special receptor kinase, bind to β-arrestin protein and in this complex lose their ability to interact with G-proteins (receptor desensitization). The complex of β 1 -adrenoreceptors with β-arrestin is absorbed by the cell by endocytosis (internalization of receptors) and is captured by endosomes and lysosomes. In the endosomes, β 1 -restin molecules are detached from receptors that re-integrate into the cell membrane; receptor sensitivity to agonists is restored (receptor resensitization). In lysosomes, the destruction of receptor molecules occurs (down-regulation) (Fig. 14).

Fig. 13. The mechanism of contraction of smooth muscles of blood vessels during stimulation of the sympathetic innervation. FLS - phospholipase C; FIF 2 - phosphatidylinositol-4,5-diphosphate; If 3 - inositol-1,4,5-triphosphate; SR - sarcoplasmic reticulum; KLCM - kinase of light chains of myosin.

Fig. 14. Desensitization and down-regulation of β-adrenergic receptors.
TO intracellular receptors corticosteroid and sex hormone receptors are included. In particular, glucocorticoid receptors are localized in the cytoplasm of cells. After glucocorticoid is combined with cytoplasmic receptors, the glucocorticoid – receptor complex penetrates the nucleus and affects the expression of various genes.
The ability of substances to bind to receptors (the tendency of substances to bind to receptors) is indicated by the term " affinity". In relation to the same receptors, the affinity of different substances can be different. To characterize affinity, use the indicator pK D is the negative logarithm of the dissociation constant, that is, the concentration of the substance at which 50% of the receptors are occupied.
Internal activity - the ability of substances to stimulate receptors; determined by pharmacological effectassociated with receptor activation.
Under normal conditions, there is no direct correlation between affinity and internal activity. A substance can occupy all receptors and cause a weak effect, and vice versa, a substance can occupy 10% of the receptors and cause the maximum effect for this system.
Agonists - substances with affinity and internal activity.
Complete agonists possess affinity and maximum internal activity (they can cause the maximum effect for a given system), even if they occupy part of specific receptors.
Partial (partial) agonists possess affinity and less than maximum internal activity (they can cause only less than maximum effects, even if they occupy 100% of specific receptors).
Antagonistspossess affinity, but do not possess internal activity and interfere with the action of full or partial agonists (displace agonists from communication with receptors).
If the action of the antagonist is eliminated by increasing the dose of the agonist, such antagonism is called competitive.
Partial agonists can be antagonists of full agonists. In the absence of a complete agonist, a partial agonist stimulates receptors and causes a weak effect. When interacting with a complete agonist, a partial agonist occupies receptors and inhibits the action of a complete agonist. In this case, the effect of a complete agonist is weakened.
For example, pindolol, a partial β-adrenoreceptor agonist, causes weak tachycardia in the absence of effects of sympathetic innervation on the heart. But with an increase in the tone of sympathetic innervation, pindolol acts as a real β-blocker and causes bradycardia. This is due to the fact that the partial agonist pindolol weakens the effect of the mediator norepinephrine, which is a complete agonist with respect to β-adrenergic receptors of the heart.
Antagonist agonists - substances that act differently on subtypes of the same receptors: they stimulate some receptor subtypes, and block others. For example, the narcotic analgesic nalbuphine has different effects on opioid receptor subtypes. Nalbuphine stimulates κ receptors (and therefore reduces pain sensitivity), and μ receptors blocks (and therefore less dangerous in terms of drug dependence).
An example of the effect of substances on enzymes there may be the action of anticholinesterase agents that block acetylcholinesterase (an enzyme that breaks down acetylcholine) and thus enhances and lengthens the action of acetylcholine.
Known medicinal substances that stimulate or block ion channels cell membranes, that is, channels that selectively conduct Na +, K +, Ca 2+ ions (sodium, potassium, calcium channels), etc. For example:
Local anesthetics block Na + channels;
Class I antiarrhythmic drugs (quinidine, lidocaine) block Na + channels;
Minoxidil activates K + channels;
Hypoglycemic agents from the group of sulfonylurea derivatives block ATP-dependent K + channels;
Verapamil, nifedipine block Ca 2+ channels.
An example of the effect of substances on transport systems there may be an action:
Reserpine (blocks vesicular uptake of dopamine and norepinephrine);
Cardiac glycosides (inhibit Na + / K + -ATPase);
Tricyclic antidepressants (block reverse neuronal uptake of norepinephrine and serotonin);
Proton pump blockers (omeprazole, etc.).
Other mechanisms of action are possible. For example, the diuretic mannitol increases diuresis by increasing the osmotic pressure in the renal tubules. The anti-atherosclerotic agent - colestipol - binds (sequestrates) bile acids, prevents their absorption in the intestine, and therefore the formation of bile acids from cholesterol in the liver is activated and the level of cholesterol in hepatocytes decreases.
The mechanisms of action of various drugs have been studied to varying degrees. In the process of studying them, ideas about the mechanisms of action can not only become more complicated, but also change significantly.
The concept of " action localization"Means the primary place (s) of action of certain medicinal substances. For example, cardiac glycosides act mainly on the heart.
To the concept of " types of action»Relate to local and general (resorptive) actions, reflex action, primary and secondary action, direct and indirect action.
An example of a local action may be the action of local anesthetics.
Most drugs have a general (resorptive) effect, which usually develops after absorption (resorption) of a substance in the blood and its distribution in the body.
Both with local and with resorptive actions, substances can excite various sensitive receptors and cause reflex reactions.
The main effect of a medicinal substance is its effects, which are used in each case. All other effects are evaluated as manifestations of side effects.
Medicinal substances can exert on certain organs direct action. In addition, the effect of drugs can be indirect. For example, cardiac glycosides have a direct effect on the heart, but, improving the functioning of the heart, these substances increase blood supply and the functions of other organs (indirect effect).