กลไกการออกฤทธิ์ สารสมุนไพร - สิ่งเหล่านี้เป็นวิธีที่สารก่อให้เกิดผลทางเภสัชวิทยา กลไกหลักของการกระทำของยาเสพติดรวมถึง:
กายภาพ
กลไกของปฏิกิริยาเคมีโดยตรง
เมมเบรน (เคมี - ฟิสิกส์)
เอนไซม์ (ชีวเคมี)
ตัวรับ
กลไกทางกายภาพของการกระทำ การกระทำของสารยาเกี่ยวข้องกับคุณสมบัติทางกายภาพ ตัวอย่างเช่นถ่านกัมมันต์ถูกประมวลผลเป็นพิเศษและดังนั้นจึงมีพื้นผิวสูง สิ่งนี้ทำให้เขาสามารถดูดซับก๊าซอัลคาลอยด์สารพิษ ฯลฯ
ปฏิกิริยาเคมีโดยตรง นี่เป็นกลไกที่ค่อนข้างหายากของการกระทำของยาเสพติดสาระสำคัญคือยาเสพติดมีปฏิสัมพันธ์โดยตรงกับโมเลกุลหรือไอออนในร่างกาย กลไกการออกฤทธิ์เช่นนั้นถูกครอบครองโดยหน่วยยาที่เป็นยากลุ่มของยาแก้พิษ ในกรณีที่เป็นพิษกับสารพิษ thiol รวมถึงเกลือของโลหะหนักหน่วย unitiol เข้าสู่ปฏิกิริยาทางเคมีโดยตรงกับพวกเขาส่งผลให้เกิดการก่อตัวของคอมเพล็กซ์ปลอดสารพิษที่ถูกขับออกมาในปัสสาวะ ดังนั้นยาลดกรดก็ทำหน้าที่เช่นกันซึ่งทำปฏิกิริยาโดยตรงกับกรดไฮโดรคลอริกลดความเป็นกรดของน้ำย่อย
มีลักษณะเป็นพังผืด (ทางกายภาพและทางเคมี) กลไก มันมีความสัมพันธ์กับผลกระทบของยาเสพติดที่มีต่อไอออนไอออน (Na +, K +, Cl - และอื่น ๆ ) ซึ่งเป็นตัวกำหนดศักย์ไฟฟ้าของสเตมเบรน ตามกลไกนี้ยาชายา antiarrhythmic ยาชาเฉพาะที่ ฯลฯ
เอนไซม์ (ชีวเคมี) กลไก กลไกนี้ถูกกำหนดโดยความสามารถของยาบางชนิดในการออกฤทธิ์กระตุ้นหรือยับยั้งเอนไซม์ คลังแสงของยาที่มีกลไกของการกระทำนั้นกว้างมาก ตัวอย่างเช่นยาเสพติด anticholinesterase, monoamine oxidase inhibitors, ปั๊มโปรตอน ฯลฯ
กลไกตัวรับ ในร่างกายมนุษย์มีสารออกฤทธิ์ทางชีวภาพที่เฉพาะเจาะจง (ผู้ไกล่เกลี่ย) ที่ทำปฏิกิริยากับตัวรับและเปลี่ยนการทำงานของอวัยวะหรือเนื้อเยื่อต่างๆของร่างกาย
ตัวรับคือโครงสร้างโมเลกุลขนาดใหญ่ที่มีความไวต่อการเลือกสารเคมีบางชนิด กับปฏิกิริยาของยากับผู้รับการเปลี่ยนแปลงทางชีวเคมีและสรีรวิทยาในร่างกายเกิดขึ้นพร้อมกับผลทางคลินิกอย่างใดอย่างหนึ่ง
ผู้ไกล่เกลี่ยและยาเสพติดที่เปิดใช้งานตัวรับและก่อให้เกิดผลกระทบทางชีวภาพที่เรียกว่า agonists. สารสมุนไพรที่ผูกกับผู้รับ แต่ไม่ทำให้เกิดการกระตุ้นและผลกระทบทางชีวภาพลดหรือกำจัดผลกระทบของ agonists เรียกว่า คู่อริ. จัดสรรด้วย ศัตรูตัวเอก - สารที่ทำหน้าที่แตกต่างกับชนิดย่อยของตัวรับเดียวกัน: พวกมันจะกระตุ้นชนิดย่อยของตัวรับและปิดกั้นตัวอื่น ๆ ยกตัวอย่างเช่นยาเสพติดยาแก้ปวด nalbuphine ช่วยกระตุ้นผู้รับ opioid คัปปา (ดังนั้นลดความไวความเจ็บปวด) และบล็อก opioid mu ผู้รับ (ดังนั้นจึงเป็นอันตรายน้อยลงในแง่ของการพึ่งพายาเสพติด)
ความสามารถของสารในการผูกกับตัวรับนั้นถูกอ้างถึงโดยคำว่า "ความสัมพันธ์" ในความสัมพันธ์กับตัวรับเดียวกันความสัมพันธ์ของสารที่แตกต่างกันอาจแตกต่างกัน
ตัวรับประเภทต่อไปนี้มีความโดดเด่น:
ตัวรับเมมเบรนพลาสม่า:
ประเภทช่องทาง: ตัวรับสัญญาณ cholinergic ชนิด N, ตัวรับแบบกล้ามเนื้อ H-cholinergic, ตัวรับ GABA;
ตัวรับ G- โปรตีน: ตัวรับα-และβ-adrenergic, M 3 -cholinoreceptors;
ตัวรับชนิดรวม: ไม่มีตัวรับ
cytosolic
ยล
เภสัชศาสตร์เป็นส่วนหนึ่ง เภสัชวิทยาทั่วไปศึกษาคุณสมบัติของการออกฤทธิ์ของยาในร่างกาย คือการศึกษาเภสัชศาสตร์:
- กลไกการออกฤทธิ์ของยา
- ปลาย ผลทางเภสัชวิทยา;
- การพึ่งพาการกระทำของยาเสพติดในเงื่อนไขต่าง ๆ ;
- ผลกระทบของยาเสพติดที่มีการบริหารซ้ำ;
- การกระทำแบบผสมผสานของยาเสพติด
- ความไม่ลงรอยกันของยา
- ผลข้างเคียงของยาเสพติด
กลไกการออกฤทธิ์ ยาเสพติด
กลไกการออกฤทธิ์ของยาเป็นวิธีการที่สารก่อให้เกิดผลทางเภสัชวิทยา กลไกหลักของการกระทำของยาเสพติดรวมถึง:
- กายภาพ
- กลไกของปฏิกิริยาเคมีโดยตรง
- เมมเบรน (เคมี - ฟิสิกส์)
- เอนไซม์ (ชีวเคมี)
- ตัวรับ
กลไกทางกายภาพของการกระทำ การกระทำของสารยาเกี่ยวข้องกับคุณสมบัติทางกายภาพ ตัวอย่างเช่นถ่านกัมมันต์ถูกประมวลผลเป็นพิเศษและดังนั้นจึงมีพื้นผิวสูง สิ่งนี้ทำให้เขาสามารถดูดซับก๊าซอัลคาลอยด์สารพิษ ฯลฯ
ปฏิกิริยาเคมีโดยตรง นี่เป็นกลไกที่ค่อนข้างหายากของการกระทำของยาเสพติดสาระสำคัญคือยาเสพติดมีปฏิสัมพันธ์โดยตรงกับโมเลกุลหรือไอออนในร่างกาย กลไกการออกฤทธิ์เช่นนั้นถูกครอบครองโดยหน่วยยาที่เป็นยากลุ่มของยาแก้พิษ ในกรณีที่เป็นพิษกับสารพิษ thiol รวมถึงเกลือของโลหะหนักหน่วย unitiol เข้าสู่ปฏิกิริยาทางเคมีโดยตรงกับพวกเขาส่งผลให้เกิดการก่อตัวของคอมเพล็กซ์ปลอดสารพิษที่ถูกขับออกมาในปัสสาวะ ดังนั้นยาลดกรดก็ทำหน้าที่เช่นกันซึ่งทำปฏิกิริยาโดยตรงกับกรดไฮโดรคลอริกลดความเป็นกรดของน้ำย่อย
มีลักษณะเป็นพังผืด (ทางกายภาพและทางเคมี) กลไก มันมีความสัมพันธ์กับผลกระทบของยาเสพติดที่มีต่อไอออนไอออน (Na +, K +, Cl - และอื่น ๆ ) ซึ่งเป็นตัวกำหนดศักย์ไฟฟ้าของสเตมเบรน ตามกลไกนี้ยาชายา antiarrhythmic ยาชาเฉพาะที่ ฯลฯ
เอนไซม์ (ชีวเคมี) กลไก กลไกนี้ถูกกำหนดโดยความสามารถของยาบางชนิดในการออกฤทธิ์กระตุ้นหรือยับยั้งเอนไซม์ คลังแสงของยาที่มีกลไกของการกระทำนั้นกว้างมาก ตัวอย่างเช่นยาเสพติด anticholinesterase, monoamine oxidase inhibitors, ปั๊มโปรตอน ฯลฯ
กลไกตัวรับ ในร่างกายมนุษย์มีสารออกฤทธิ์ทางชีวภาพที่เฉพาะเจาะจง (ผู้ไกล่เกลี่ย) ที่ทำปฏิกิริยากับตัวรับและเปลี่ยนการทำงานของอวัยวะหรือเนื้อเยื่อต่างๆของร่างกาย
ตัวรับคือโครงสร้างโมเลกุลขนาดใหญ่ที่มีความไวต่อการเลือกสารเคมีบางชนิด กับปฏิกิริยาของยากับผู้รับการเปลี่ยนแปลงทางชีวเคมีและสรีรวิทยาในร่างกายเกิดขึ้นพร้อมกับผลทางคลินิกอย่างใดอย่างหนึ่ง
ผู้ไกล่เกลี่ยและยาเสพติดที่เปิดใช้งานตัวรับและก่อให้เกิดผลกระทบทางชีวภาพที่เรียกว่า agonists. สารสมุนไพรที่ผูกกับผู้รับ แต่ไม่ทำให้เกิดการกระตุ้นและผลกระทบทางชีวภาพลดหรือกำจัดผลกระทบของ agonists เรียกว่า คู่อริ. จัดสรรด้วย ศัตรูตัวเอก - สารที่ทำหน้าที่แตกต่างกับชนิดย่อยของตัวรับเดียวกัน: พวกมันจะกระตุ้นชนิดย่อยของตัวรับและปิดกั้นตัวอื่น ๆ ยกตัวอย่างเช่นยาเสพติดยาแก้ปวด nalbuphine ช่วยกระตุ้นผู้รับ opioid คัปปา (ดังนั้นลดความไวความเจ็บปวด) และบล็อก opioid mu ผู้รับ (ดังนั้นจึงเป็นอันตรายน้อยลงในแง่ของการพึ่งพายาเสพติด)
ความสามารถของสารในการผูกกับตัวรับนั้นถูกอ้างถึงโดยคำว่า "ความสัมพันธ์" ในความสัมพันธ์กับตัวรับเดียวกันความสัมพันธ์ของสารที่แตกต่างกันอาจแตกต่างกัน
ตัวรับประเภทต่อไปนี้มีความโดดเด่น:
- ตัวรับเมมเบรนพลาสม่า:
- ประเภทช่องทาง: ตัวรับสัญญาณ cholinergic ชนิด N, ตัวรับแบบกล้ามเนื้อ H-cholinergic, ตัวรับ GABA;
- ตัวรับ G- โปรตีน: ตัวรับα-และβ-adrenergic, M 3 -cholinoreceptors;
- ตัวรับชนิดรวม: ไม่มีตัวรับ
- cytosolic
- ยล
- นิวเคลียร์
ตัวรับเมมเบรนพลาสม่า
ตัวรับประเภทช่อง
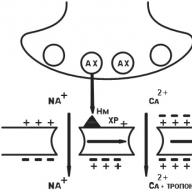
ตัวรับ n-cholinergic ของประเภทเส้นประสาท (ระบบประสาทส่วนกลางปมประสาทอัตโนมัติ, โซน sinocarotid, เนื้อเยื่อต่อมหมวกไต chromaffin) หลังจากการจับ acetylcholine (AX) กับตัวรับ Hn-cholinergic, Na + จะเปิดช่องทางและ Na พุ่งเข้าสู่เซลล์โดยมีประจุเป็นบวก เมมเบรน postsynaptic เป็น depolarized มีความเป็นไปได้ที่จะเคลื่อนที่ไปตามเยื่อหุ้มเซลล์ของเซลล์ประสาทซึ่งเปิดช่องสัญญาณ Na + ที่ขึ้นกับกระแสไฟฟ้า แรงกระตุ้นเส้นประสาทเกิดขึ้นในเส้นใย postganglionic (รูปที่ 6)
มะเดื่อ 6. ตัวรับ n-cholinergic
N m - ตัวรับ cholinergic ประเภทกล้ามเนื้อ เยื่อหุ้มเซลล์กล้ามเนื้อโครงร่าง. กระบวนการเริ่มต้นคล้ายกัน แต่เปิดช่องทาง Ca ++ ทางไฟฟ้าได้ Ca ++ ไอออนเข้าสู่เส้นใยกล้ามเนื้อ Ca ++ ถูกปลดปล่อยจาก reticulum sarcoplasmic ระดับของ Ca ++ เพิ่มขึ้นซึ่งจะทำให้กล้ามเนื้อหดตัว (รูปที่ 7)

มะเดื่อ 7. N m-cholinergic receptor
ตัวรับ GABA เหล่านี้คือตัวรับสำหรับกรดγ-aminobutyric (GABA) GABA โต้ตอบกับตัวรับ GABA ในโครงสร้างที่มีช่องคลอไรด์ เป็นผลมาจากการกระตุ้นตัวรับช่องเปิดและคลอรีนไอออน (Cl -) เข้าสู่เซลล์ได้อย่างอิสระ การเพิ่มขึ้นของความเข้มข้นของคลอรีนไอออนภายในเซลล์จะนำไปสู่การเพิ่มจำนวนมากของเยื่อหุ้มเซลล์และลดการทำงานของเซลล์ประสาท มันยากกว่าที่จะกระตุ้นเซลล์เช่นนี้ (รูปที่ 8)

มะเดื่อ 8. ตัวรับ GABA:
GABA-R - ตัวรับ GABA, BD-R - ตัวรับ benzodiazepine, BR - ตัวรับแบบ barbiturate
ผู้รับที่เกี่ยวข้องกับ โปรตีนจี
G-proteins เช่นโปรตีน GTP-binding (guanosine triphosphate-binding) โปรตีนถูกแปลเป็นภาษาท้องถิ่นในเยื่อหุ้มเซลล์และประกอบด้วยα-, β-และγ-subunits พวกเขา (G-proteins) ควบคุมกิจกรรมของเอฟเฟกต์เฉพาะ (ผู้ส่งสารทันที, ตัวกลางรอง) สารเหล่านี้สามารถเป็นเอนไซม์ (adenylate cyclase, phospholipase); ช่องทางสำหรับโพแทสเซียมแคลเซียมโซเดียม โปรตีนขนส่งบางชนิด แต่ละเซลล์สามารถมี G-โปรตีนจำนวนมากแต่ละเซลล์ควบคุมกิจกรรมของผู้ส่งสารต่าง ๆ ในขณะที่เปลี่ยนการทำงานของเซลล์
M 3 -cholinergic receptor (เยื่อหุ้มกล้ามเนื้อเรียบ (MMCs) และเซลล์ต่อม exocrine) Acetylcholine ช่วยกระตุ้น M 3 -XR ที่จับกับโปรตีน G เปิดใช้งาน Phospholipase-C (FLS) ซึ่งกระตุ้นการแตกตัวของ PIDP (phosphatidylinositol diphosphate) เข้าสู่ ITP (inositol triphosphate) และ DAG (diacylglycerol) ITF เข้าสู่ไซโตพลาสซึมของ MMC ปล่อย Ca ++ จาก cavelles .

มะเดื่อ 9. ตัวรับ M3-cholinergic
Ca ++ ผูกกับคาโลดีลินเปิดใช้งาน myosine kinase (MK) ซึ่งเป็นตัวเร่งปฏิกิริยาฟอสโฟรีเลชั่นของโซ่แสง myosin ซึ่งนำไปสู่การหดตัวของเซลล์ (รูปที่ 9) ในทำนองเดียวกันแรงกระตุ้นจะถูกส่งไปที่ synapses ของต่อมหลั่ง
Norepinephrine ช่วยกระตุ้น α 1 -adrenoreceptceptorโดยเริ่มห่วงโซ่ของเหตุการณ์ต่อไปนี้:
Norepinephrine (HA) →α 1 -adrenoreceptor →การกระตุ้นการทำงานของα-subunit ของ G s-protein →การกระตุ้น PLL →ความแตกแยกของ FIDF →การเพิ่มความเข้มข้นของ ITF →การเพิ่มขึ้นของความเข้มข้นของ ITF →ในความเข้มข้นของ Ca 2+ ในเซลล์→ Ca 2+ myosin → myosin โต้ตอบกับ actin →การลดลงของ MMC ที่พัฒนา (รูปที่ 10)

มะเดื่อ 10. α 1 -adrenoreceptor
ข 1 ตัวรับ(รูปที่ 11) Norepinephrine →เปิดใช้งาน b 1 -AP →การกระตุ้นของα-subunit ของ G-protein →การกระตุ้น AC →เพิ่มการผลิต cAMP จาก ATP →เพิ่มความเข้มข้นของ cAMP ใน cardiomyocyte →การกระตุ้นของ kinases โปรตีน→ phosphorylation ของแคลเซียมแชนแนลโปรตีน→เพิ่มขึ้นในช่อง Ca และ 2 2+ ในเซลล์→การเพิ่มขึ้นของแรงบีบตัวของหัวใจ

มะเดื่อ 11 b 1 ตัวรับ
ข 2 ตัวรับ(รูปที่ 12) ON → b 2 -AP →การเปิดใช้งานของα-subunit ของ G-protein →การกระตุ้น AC →การก่อตัวของแคมป์ที่เพิ่มขึ้น→ kinase โปรตีนกระตุ้น→ kinase ที่เร่งปฏิกิริยา phosphorylation ของ myosine kinase นั้นจะถูกแยกออกในขณะที่กิจกรรมหลังหายไป→ Myosin phosphorylation
กฎระเบียบของการเปิดตัวของ HA จากปลายประสาทจะดำเนินการโดยสารสื่อประสาทตัวเองในการกระตุ้นของเมมเบรน presynaptic α 2 -AP การปล่อย HA ลดลง

มะเดื่อ 12 b 2 ตัวรับ
ตัวรับชนิดเชิงบูรณาการ
เหล่านี้คือตัวรับที่เป็นโปรตีนที่แทรกซึมผ่านเยื่อหุ้มเซลล์ ในกรณีนี้ส่วนนอกของโปรตีนมีบทบาทตัวรับในขณะที่ส่วนด้านในมีบทบาทในการเร่งปฏิกิริยา (รูปที่ 13)

มะเดื่อ 13 ตัวรับชนิดเชิงบูรณาการ
ตัวรับ Cytosolic
ภายใต้สภาวะทางสรีรวิทยาผู้รับเช่นนั้นจะทำหน้าที่ผูกฮอร์โมนสเตียรอยด์ (ฮอร์โมนเพศกลูโคคอร์ติคอยด์) สารเหล่านี้เข้าสู่เซลล์และผูกเข้ากับตัวรับ cytosolic ที่นั่น คอมเพล็กซ์นี้แทรกซึมนิวเคลียสและเปลี่ยนการทำงานของจีโนมที่นั่น เป็นผลให้การสังเคราะห์โปรตีนในเซลล์เปลี่ยนไป (รูปที่ 14)
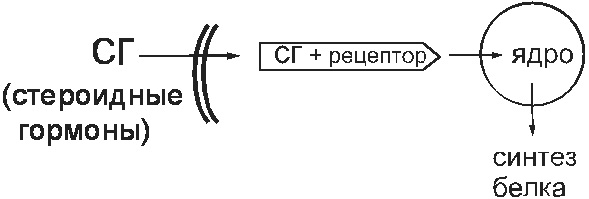
มะเดื่อ 14 ตัวรับ Cytosolic
ตัวรับไมโตคอนเดรีย
ในไมโตคอนเดรียยังมีตัวรับซึ่งสารสมุนไพรมีปฏิกิริยาเช่น triiodothyronine hydrochloride ซึ่งเป็นแอนะล็อกของฮอร์โมนธรรมชาติ T 3 จากการโต้ตอบนี้การสังเคราะห์ ATP จะเพิ่มขึ้น
ตัวรับนิวเคลียร์
T 3 แทรกซึมเข้าไปในนิวเคลียสและมีปฏิสัมพันธ์กับตัวรับประเภทนี้ เป็นผลให้การทำงานของการเปลี่ยนแปลงจีโนมและโปรตีนใหม่ถูกสังเคราะห์
ผลทางเภสัชวิทยาสุดท้าย (ตาม Vershinin)
แม้จะมียาเสพติดมากมายการเปลี่ยนแปลงที่เกิดจากพวกมันในร่างกายเป็นประเภทเดียวกัน (รูปที่ 15) ผลกระทบของยาเสพติดใด ๆ ในอวัยวะสามารถลดลงถึงห้าผลทางเภสัชวิทยาหลัก (ตาม N.V. Vershinin):
- การปลอบใจ - ลดการเข้าสู่ภาวะปกติของการทำงานของอวัยวะเพิ่มขึ้น (การใช้ยาระงับประสาท)
- การกดขี่ - ลดลงต่ำกว่าบรรทัดฐานของการทำงานของร่างกาย (ใช้ยาระงับความรู้สึก)
- อัมพาต - หยุดการทำงานของอวัยวะลดลง (ระบบทางเดินหายใจในกรณียาเกินขนาดของยาแก้ปวดยาเสพติด)
- ปรับสีขึ้น - เพิ่มความแข็งแรงของฟังก์ชั่นที่ลดลงสู่ปกติ
- กระตุ้น - เพิ่มการทำงานของอวัยวะเกินกว่ามาตรฐาน (การใช้ยาขับปัสสาวะในกรณีที่เป็นพิษ, ยาขับเสมหะ)
มะเดื่อ 15. ผลทางเภสัชวิทยาสุดท้าย

ประเภทของการกระทำของยาเสพติด
- สิ่งสำคัญคือรอง
สิ่งสำคัญ การกระทำคือสิ่งที่อยู่ภายใต้การบริหารการรักษาหรือป้องกันโรคของยาเสพติด ด้าน - ไม่พึงประสงค์อันตรายสำหรับการกระทำของผู้ป่วยของยาเสพติด
- ย้อนกลับไม่สามารถย้อนกลับได้
เมื่ออยู่ในร่างกายสารยาจะทำปฏิกิริยากับเซลล์เหล่านั้นซึ่งมีสารชีวภาพที่สามารถทำปฏิกิริยากับสารนี้ได้ ปฏิกิริยานี้ขึ้นอยู่กับโครงสร้างทางเคมีของยา การรวมตัวกันของสารยากับสารตั้งต้นที่เหมาะสมคือ ย้อนกลับ ถ้าพวกเขา (สารตั้งต้นและยาเสพติด) ผูกกันในขณะที่
ในบางกรณีเป้าหมายการรักษาต้องการ กลับไม่ได้ การปิดโครงสร้างจากฟังก์ชัน ตัวอย่างนี้ใช้กับยาต้านจุลชีพส่วนใหญ่สารต้านมะเร็งที่สามารถสร้างพันธะที่แข็งแกร่ง (โควาเลนต์) ที่มีองค์ประกอบของเซลล์ DNA helix ("crosslinking") หรือเอนไซม์แบคทีเรียซึ่งเป็นผลมาจากเซลล์สูญเสียความสามารถในการทำซ้ำ
- โดยตรง, ทางอ้อม (ทางอ้อม)
โดยตรง การกระทำที่แสดงให้เห็นว่าผลการรักษาเกิดจากการทำงานร่วมกันโดยตรงของยาเสพติดกับ biosubstrate ของอวัยวะที่เป็นโรคและนำไปสู่การเปลี่ยนแปลงบางอย่างโดยตรง หากการทำงานของอวัยวะ (ระบบ) เปลี่ยนเป็นครั้งที่สองเป็นผล อิทธิพลโดยตรง ยาในอวัยวะอื่นระบบอื่นการกระทำนี้เรียกว่าทางอ้อม (ทางอ้อม) glycosides หัวใจปรับปรุงการหดตัวของกล้ามเนื้อหัวใจ (ผลกระทบโดยตรง) และเป็นผลให้ปรับปรุงการไหลเวียนโลหิตในร่างกายซึ่งจะมาพร้อมกับการปรับปรุงใน diuresis (ผลทางอ้อม)
กรณีพิเศษของการกระทำทางอ้อมคือ สะท้อน การกระทำ ตัวอย่างเช่นการขยายตัวของหลอดเลือดและการปรับปรุงเนื้อเยื่อ trophic อันเป็นผลมาจากการระคายเคืองที่ปลายประสาทสัมผัสของผิวหนัง
- เลือกไม่เลือก
การกระทำที่เลือก คือผลกระทบของปริมาณยาที่ใช้รักษาต่อตัวรับเฉพาะ ตัวอย่างเช่นผลของ salbutamol ต่อβ 2-adrenoreceptors มันควรจะเป็นพาหะในใจว่าการเลือกของยาเสพติดมีความสัมพันธ์กับปริมาณที่เพิ่มขึ้นก็จะหายไป
- ในพื้นที่ resorptive
ในประเทศ ผลกระทบของยาเสพติดจะดำเนินการก่อนที่จะถูกดูดซึมเข้าสู่กระแสเลือด (ตัวอย่างเช่นครีม)
resorptive การกระทำ (ระบบ) พัฒนาหลังจากการดูดซึมของยาเข้าสู่เลือด ยาเสพติดส่วนใหญ่มีผลกระทบนี้
ในกรณีส่วนใหญ่สำหรับสารยา (ลิแกนด์) ที่จะออกฤทธิ์มันจะต้องพบส่วนประกอบเฉพาะในตัวรับเป้าหมายโครงสร้างโมเลกุลที่เป็นโปรตีนกรดนิวคลีอิกน้อยกว่าไขมันหรือการกำหนดค่าอื่น ๆ ที่อยู่ภายในหรือบนพื้นผิวของเซลล์ ด้วยการโต้ตอบเริ่มต้นสายโซ่ของกระบวนการทางชีวเคมีและเคมี - ฟิสิกส์ที่นำไปสู่
มีตัวรับเมมเบรนสองประเภท - ช่องไอออนและตัวรับที่เกี่ยวข้องกับโปรตีนจี ตัวอย่างเช่นช่องโซเดียมเป็นลักษณะของ adetylcholine และยาที่คล้ายกัน Acetylcholine ทำปฏิกิริยากับโปรตีนแชนเนลทำให้เกิดการเปลี่ยนแปลงในโครงสร้างซึ่งส่งผลต่อการเปิดช่องทางและการแทรกซึมของไอออนโซเดียมในเซลล์ กระบวนการนี้ต้องใช้ความตื่นเต้นตื่นเต้น สารบางอย่างที่ทำปฏิกิริยากับโปรตีนของโซเดียมแชนเนลป้องกันการเปิดดังนั้นการปิดกั้นการส่งผ่านของการกระตุ้นประสาท
G- โปรตีนที่เรียกว่าถูกแนบไปกับส่วนด้านในของพลาสมาเมมเบรนของเซลล์ซึ่งช่วยให้แน่ใจว่าการประสานของกระบวนการปฏิสัมพันธ์ของสารยาด้วยการเปิดใช้งานพร้อมกันของโปรตีนเป้าหมายเซลล์ภายในที่สอดคล้องกัน ดังที่แสดงในรูปโมเลกุลของยาจะทำปฏิกิริยากับตัวรับ (P) ที่ผิวด้านนอกของเมมเบรนซึ่งทำให้เกิดการเปลี่ยนแปลงโครงสร้างของโปรตีนตัวรับ ด้วยเหตุนี้ G-protein จึงเปลี่ยนโครงสร้างเชิงพื้นที่จึงย้ายไปอยู่ในระนาบของเมมเบรนเป็นเอ็นไซม์ที่อยู่ในสถานะไม่ได้ใช้งานภายในเซลล์ การทำงานร่วมกันของ G-โปรตีนกับเอนไซม์ (T) กำหนดเปิดใช้งานของพวกเขา (LV / P / T) Norepinephrine, dopamine และลิแกนด์อื่น ๆ มีปฏิกิริยาเฉพาะกับตัวรับที่เกี่ยวข้องกับ G-protein มันควรจะสังเกตว่า acetylcholine สามารถโต้ตอบไม่เพียงกับโปรตีนช่อง แต่ยังมีตัวรับที่เกี่ยวข้องกับโปรตีน G
สำหรับการทำงานร่วมกันระหว่างแกนด์และตัวรับ bioreceptor มันเป็นสิ่งจำเป็นที่พวกเขามี complementarity นั่นคือระหว่างพวกเขาจะต้องมีความสัมพันธ์หรือความสัมพันธ์ (การติดต่อกันของขนาดการกำหนดค่าเชิงพื้นที่การปรากฏตัวของค่าตรงกันข้าม) ยกตัวอย่างเช่นประจุลบของตัวรับจะต้องสอดคล้องกับประจุบวกของแกนด์ภายนอกและอนุมูลที่ไม่ใช่ขั้วของสารสามารถจับกับไซต์ที่ไม่ชอบน้ำของตัวรับ
ในคุณสมบัติทางเคมีกายภาพของสารยาที่มีผลต่อการโต้ตอบกับผู้รับเราควรแยกขนาดของโมเลกุลขึ้นอยู่กับว่าสารสามารถโต้ตอบกับตัวรับทั้งหมดหรือส่วนประกอบ จลนพลศาสตร์ของการแทรกซึมผ่านเยื่อหุ้มชีวภาพยังขึ้นอยู่กับขนาดของโมเลกุลยาด้วย โดยทั่วไปเมื่อขนาดของโมเลกุลเพิ่มขึ้นความยืดหยุ่นและความเป็นไปได้ของการก่อตัวของพันธบัตรแวนเดอร์วาวาลส์ด้วยการเพิ่มขนาดโมเลกุลใหญ่ขึ้น นอกจากนี้ stereochemistry ของโมเลกุลยานั้นมีความสำคัญ กิจกรรมทางเภสัชวิทยาขึ้นอยู่กับรูปแบบไอโซเมอร์ของสารยา และสิ่งหนึ่งที่ต้องจำไว้คือความแข็งของโครงสร้างของโมเลกุลของตัวรับความแตกต่างในการกระทำของสเตอริโอไอโซเมอร์
ปฏิกิริยาของสารเสพติด - ตัวรับเกิดจากพันธะระหว่างโมเลกุล. ในขั้นต้นสารถูกดึงดูดไปยังตัวรับโดยกองกำลังไฟฟ้าสถิตและในการปรากฏตัวของ complementarity มันจะสร้างพันธะกับตัวรับโดยใช้ปฏิกิริยาทางกายภาพและทางเคมีกายภาพ (โดยทั่วไปสำหรับยาที่ถูกขับออกจากร่างกายในรูปแบบไม่เปลี่ยนแปลงหรือไม่เปลี่ยนแปลง) ผ่านการเปลี่ยนแปลงทางเคมีในร่างกาย) กองกำลัง van der Waals ที่อ่อนแอที่สุดมีส่วนร่วมในการกำหนดความจำเพาะของปฏิกิริยาระหว่างยากับระบบปฏิกิริยาทางชีวเคมี พันธะไฮโดรเจนมีส่วนร่วมในกระบวนการการรับรู้และการตรึงสาร (ลิแกนด์) ไปยังโครงสร้างชีวภาพ พันธะไอออนิกเกิดขึ้นในกรณีที่สารยาประกอบด้วยกลุ่มประจุบวกหรือประจุลบและโครงสร้างตรงข้ามอยู่ในตัวรับชีวภาพ บ่อยครั้งที่พันธะไอออนิกเกิดขึ้นในระยะแรกของปฏิกิริยาทางเภสัชวิทยาระหว่างสารและตัวรับ ในกรณีดังกล่าวผลกระทบของยาเสพติดสามารถย้อนกลับได้ การก่อตัวของพันธะโควาเลนต์ประสานงานเป็นสิ่งสำคัญ ด้วยการมีส่วนร่วมของพวกเขาปฏิกิริยาของสารอัลคิลติงกับ biosubstrates เช่นเดียวกับยาและยาแก้พิษด้วยโลหะเกิดขึ้นในระหว่างการก่อตัวของสารประกอบเชิงซ้อนคีเลตที่มีความเสถียรเช่น unithiol กับสารหนูหรือ tetacin- แคลเซียมด้วยตะกั่ว การกระทำของสารดังกล่าวกลับไม่ได้
นอกจากนี้ยังมีปฏิสัมพันธ์ที่ไม่ชอบน้ำ แม้ว่าพลังงานของพันธะของมันจะมีขนาดเล็ก แต่การมีปฏิสัมพันธ์ของโซ่อะลิฟาติกยาวจำนวนมากนำไปสู่การปรากฏตัวของระบบที่เสถียร ปฏิกิริยาที่ชอบน้ำในน้ำมีบทบาทในการสร้างความมั่นคงให้กับพอลิเมอร์ชีวภาพและสร้างเยื่อหุ้มชีวภาพ
กรดอะมิโนที่ตกค้างในโมเลกุลตัวรับโปรตีนประกอบด้วยกลุ่มขั้วโลกและกลุ่มที่ไม่ใช่ขั้วที่กำหนดการก่อตัวของพันธะขั้วและขั้วที่ไม่ใช่ขั้วระหว่างพวกเขาและสารยา กลุ่มขั้วโลก (-OH, -NH, COO-, -N 3 H, \u003d O) ให้การก่อตัวของพันธะไอออนิกและไฮโดรเจนเป็นหลัก กลุ่มที่ไม่เป็นโมล (ไฮโดรเจน, เมธิล, อนุมูลอิสระ, ฯลฯ ) ก่อตัวเป็นพันธะที่ไม่เข้ากับน้ำกับสารยาที่มีน้ำหนักโมเลกุลต่ำ
ดังนั้นการโต้ตอบของยากับผู้รับที่เฉพาะเจาะจงสามารถทำได้ผ่านพันธะเคมีต่าง ๆ มีความแข็งแรงไม่เท่ากัน ดังนั้นความแข็งแรงโดยประมาณของสารคล้าย curare กับตัวรับ cholinergic สำหรับการเกิดไฟฟ้าสถิต (อิออน) คือ 5 kcal / mol, dipole-ion - 2-5 kcal / mol, dipole-dipole - 1-3 kcal / mol, พันธะไฮโดรเจน - 2-5 kcal / mol, van der Waals - 0.5 kcal / mol, hydrophobic bondet - 0.7 kcal ต่อ CH 2 กลุ่ม การลดลงของความแข็งแรงพันธะขึ้นอยู่กับระยะห่างระหว่างอะตอมของการเกิดปฏิกิริยาไฟฟ้าสถิตคือ r -2, ไดโพลอิออนคือ r -3, ไดโพลไดโพลคือ r -4, พันธะไฮโดรเจนคือ r -4, พันธะไฮโดรเจนแวนเดอร์ Waals คือ r -7 . การเชื่อมต่อชนิดนี้สามารถใช้งานไม่ได้ซึ่งจะทำให้แน่ใจได้ว่าการกระทำของยาเสพติด มีความคงทนมากขึ้นคือพันธะโควาเลนต์ซึ่งให้ผลที่ยาวนานและไม่สามารถย้อนกลับได้ของสารตัวอย่างเช่นยาต้านแอลคิลตีต ยาเสพติดส่วนใหญ่ผูกกลับได้กับผู้รับ ในกรณีนี้ตามกฎแล้วธรรมชาติของสารประกอบมีความซับซ้อนมาก: อิออน, ไดโพล - ไดโพล, แวนเดอร์วาล์ส์, ที่ไม่ชอบน้ำและพันธะประเภทอื่น ๆ สามารถมีส่วนร่วมในมันได้ซึ่งส่วนใหญ่กำหนดโดยธรรมชาติของสารและตัวรับ ด้วยตัวเอง
ความแข็งแรงของสารยึดจับกับตัวรับจะแสดงโดยคำว่า "ความสัมพันธ์". สารที่ทำกับตัวรับเดียวกันอาจมีระดับความสัมพันธ์ที่ต่างกันสำหรับพวกเขา ในกรณีนี้สารที่มีค่าความสัมพันธ์สูงจะสามารถแทนที่สารที่มีค่าความสัมพันธ์ต่ำกว่าจากสารประกอบที่มีตัวรับ ในการกำหนดสถานะสมดุลระหว่างตัวรับ "ที่ถูกครอบครอง" (DR) ตัวรับอิสระและสารอิสระ (D) จะใช้ค่าคงที่การแยกตัว (K D) ซึ่งถูกกำหนดโดยสูตรต่อไปนี้:
K D \u003d [D] * [R] / [DR]
ลอการิทึมลบของ K D (pR D) เป็นตัวบ่งชี้ความสัมพันธ์ ในการระบุลักษณะของความสัมพันธ์มักใช้ตัวบ่งชี้ pD 2 นั่นคือลอการิทึมลบของ EC 50 (ความเข้มข้นของสารที่ทำให้เกิดผล 50% ของผลกระทบสูงสุด)
ความหลากหลายของพันธะปฏิกิริยาเคมีและความแข็งแรงไม่เท่ากันหรือความสัมพันธ์ระหว่างแกนด์และตัวรับ bioreceptors อธิบายโดยโครงสร้างที่ซับซ้อนของยาที่มีอนุมูลที่มีปฏิกิริยาต่างกันและมีรูปร่างปริมาตรหลายมิติเช่นเดียวกับความซับซ้อนของกระบวนการปฏิสัมพันธ์ซึ่งมักเกิดขึ้นในหลายขั้นตอน สารเสพติดเป็นตัวรับ; การจัดกลุ่มโมเลกุล ความร้าวฉานที่ซับซ้อน
ดังนั้นเฉพาะสารที่มีความสัมพันธ์ที่เด่นชัดสำหรับตัวรับชีวภาพเท่านั้นที่สามารถทำให้เกิดผลทางเภสัชวิทยา ความรุนแรงของผลกระทบขึ้นอยู่กับความเข้มข้นของยาและจำนวนตัวรับทั้งหมด
หากสารมีกิจกรรมภายในที่เพียงพอก็จะเรียกว่า agonists. โดยกิจกรรมภายในจะเข้าใจถึงความสามารถของนัก agonists ในการทำให้เกิดผลทางชีวภาพโดยการเปลี่ยนโครงสร้างของตัวรับเช่นความสามารถของแกนด์ในการกระตุ้นตัวรับ ปรากฏการณ์นี้ถูกพิจารณาว่าเป็นความสัมพันธ์ของตัวรับ agonist กับตัวแปลงสัญญาณการแปลงสัญญาณภายนอกเป็นสัญญาณภายในเรียกว่าการแปลงสัญญาณ กระบวนการส่งสัญญาณภายในเซลล์เช่นกระบวนการหดตัวของเส้นใยกล้ามเนื้อการแบ่งเซลล์การเพิ่มความแตกต่าง ฯลฯ ขณะนี้ได้รับการยอมรับแล้วว่าเซลล์นั้นมีสารหลายอย่าง (ฮอร์โมนเปปไทด์ที่ออกฤทธิ์ทางชีวภาพนิวคลีโอไทด์สเตียรอยด์ ผู้รับที่เฉพาะเจาะจง เป็นผลมาจากการมีปฏิสัมพันธ์ของสารเหล่านี้กับผู้รับที่เฉพาะเจาะจงเหล่านี้รองผู้ส่งสาร (ตัวกลาง) จะเกิดขึ้นที่ก่อให้เกิดน้ำตกของปฏิกิริยาทางชีวเคมี
มีแนวคิดคือ " agonists บางส่วน"- สารสมุนไพรที่เมื่อถึงผู้รับจะไม่ให้ผลสูงสุด ปรากฏการณ์ที่เข้าใจยากนี้น่าจะเกิดจากการพึ่งพาอาศัยความสัมพันธ์ที่ซับซ้อนของตัวรับยาเสพติดสำหรับ traneductor ยกตัวอย่างเช่นตัวรับ opal receptor agonist nalorphine ทำหน้าที่คล้ายกับ agonist ตัวเต็มของ morphine ตัวรับเหล่านี้แม้ว่าจะอ่อนกว่ารุ่นหลัง ในเวลาเดียวกันเมื่อใช้ร่วมกัน, nalorphine ทำให้อ่อนแอหรือกำจัดผลกระทบของมอร์ฟีน; โดยเฉพาะอย่างยิ่งผลการยับยั้งมอร์ฟีนต่อการหายใจถูกกำจัด Isoprenaline เป็นตัวเอกที่แท้จริงและ prenalterol เป็นตัวเอกบางส่วนสำหรับผู้รับ ad-adrenergic ตามทฤษฎีของตัวรับตัวเอกที่แท้จริงสามารถกระตุ้นให้เกิดการตอบสนองสูงสุดแม้ว่ามันจะมีปฏิสัมพันธ์กับตัวรับเพียงบางส่วน
ผู้รับที่เฉพาะเจาะจงอาจมีเว็บไซต์ผูกพันหรือแตกต่างกันสำหรับ agonists และคู่อริ เว็บไซต์ที่มีผลผูกพันที่แตกต่างกันสำหรับผู้กระทำผิดที่แตกต่างกันเป็นไปได้ ในกรณีที่ตัวเอกและศัตรูมีไซต์ที่มีผลผูกพันเดียวกันและผลของการปิดกั้นของคู่ต่อสู้บนตัวรับจะถูกกำจัดโดยการเพิ่มความเข้มข้นของตัวเอก (ความสำเร็จสูงสุดของตัวเอกคือความสัมพันธ์ระหว่างคู่แข่งและตัวเอก) หากไซต์ที่มีผลผูกพันสำหรับตัวเอกและศัตรูนั้นแตกต่างกันความสัมพันธ์ระหว่างพวกเขาจะถูกกำหนดให้เป็นศัตรูที่ไม่ใช่คู่แข่ง เพื่อระบุลักษณะของคู่อริมักใช้ pA 2 (ลอการิทึมลบของความเข้มข้นของโมลาร์ของคู่อริซึ่งต้องเพิ่มความเข้มข้นเป็นสองเท่าเพื่อให้ได้ผลมาตรฐานของตัวเอก)
ภายใต้เงื่อนไขของสิ่งมีชีวิตทั้งหมด agonists และคู่อริทำให้เกิดการเปลี่ยนแปลงในหน้าที่ทางสรีรวิทยา ในกรณีนี้การกระทำของคู่อริจะถูกกำหนดโดยความจริงที่ว่าพวกเขายับยั้งอิทธิพลของแกนด์ธรรมชาติเฉพาะบนตัวรับที่เฉพาะเจาะจง (ตัวอย่างเช่น atropine M-cholinergic antagonist ยับยั้งการกระทำของตัวเอก acetylcholine ของพวกเขา) การเปลี่ยนแปลงที่เกี่ยวข้องโดยตรงกับการทำงานร่วมกันของสารที่มีตัวรับที่เฉพาะเจาะจงจะแสดงด้วยคำว่า "ปฏิกิริยาทางเภสัชวิทยาหลักซึ่งอาจเป็นจุดเริ่มต้นของปฏิกิริยาที่นำไปสู่การกระตุ้นหรือยับยั้งการทำงานทางสรีรวิทยาบางอย่าง"
การเปลี่ยนแปลงการทำงานของอวัยวะหรือระบบ (ตัวอย่างเช่นการเปลี่ยนแปลงความแข็งแรงและความถี่ของการหดตัวของหัวใจ, กล้ามเนื้อเรียบของอวัยวะภายใน, การหลั่งของต่อม, ความดันโลหิต, ฯลฯ ) ที่เกิดจากสารยาถูกกำหนดเป็น ผลทางเภสัชวิทยาของสารนี้. ดังนั้นสำหรับการเต้นของหัวใจ glycosides ปฏิกิริยาทางเภสัชวิทยาหลักคือการยับยั้งกิจกรรมของการขนส่ง Na +, K-ATPase ของเส้นใยกล้ามเนื้อหัวใจซึ่งถือว่าเป็นตัวรับเฉพาะที่เป็นไปได้สำหรับการเต้นของหัวใจ glycosides ในเรื่องนี้การเข้าสู่เส้นใยกล้ามเนื้อ K + และออกจากเส้นใย Na + จะหยุดชะงักและเนื้อหา Ca2 + ในไซโตพลาสซึมเพิ่มขึ้นซึ่งส่งเสริมการทำงานร่วมกันของ actin และ myosin ผลของการเปลี่ยนแปลงเหล่านี้คือการเพิ่มขึ้นของอัตราการเต้นของหัวใจซึ่งเป็นผลทางเภสัชวิทยาหลักของการเต้นของหัวใจ glycosides
การเปิดรับแสงเป็นเวลานานของตัวรับที่เฉพาะเจาะจงกับผู้ชำนาญการมักจะมาพร้อมกับการลดลงของความไวของพวกเขา หลังอาจเกี่ยวข้องกับการเปลี่ยนแปลงในการรับลดจำนวน (ความหนาแน่น) หรือการหยุดชะงักในกระบวนการที่เป็นไปตามการกระตุ้นของผู้รับ ในกรณีนี้ผลทางเภสัชวิทยาของผู้กระทำผิดมีความชัดเจนน้อยลง
ดังนั้นผลทางเภสัชวิทยาของยาส่วนใหญ่จึงสัมพันธ์กับผลกระทบต่อตัวรับเฉพาะที่เกี่ยวข้อง
สารที่มีความเกี่ยวข้องสูงสำหรับตัวรับ Bioreceptor และกิจกรรมภายในที่ต่ำเรียกว่าคู่อริหรือตัวบล็อกเนื่องจากมันไม่ทำให้เกิดการเปลี่ยนแปลงโครงสร้างของตัวรับ bioreceptor ยับยั้งการทำงานร่วมกันของลิแกนด์ภายนอกและ / หรือ agonist ภายนอก มีสิ่งที่เรียกว่า "ตัวรับผลทุติยภูมิหรือตัวใบ้ซึ่งสารตัวนี้ผูกมัด แต่ไม่มีผลทางเภสัชวิทยา ตัวรับ "โง่" ดังกล่าวมักมีอยู่ในโปรตีนและพลาสมาในเลือด (แต่สามารถพบได้ในเนื้อเยื่อ) การเชื่อมต่อกับตัวรับ "ปิดเสียง" จะนำไปสู่การลดความเข้มข้นของสารเสพติดฟรีและดังนั้นจึงลดลงในผลการรักษา
ทฤษฎีสมัยใหม่จำนวนมากอธิบายกลไกของการมีปฏิสัมพันธ์ระหว่างลิแกนด์และตัวรับสถานะของตัวรับเองการขาดสัดส่วนระหว่างจำนวนตัวรับที่ถูกยึดครองและปฏิกิริยาสุดท้ายการเปลี่ยนแปลงประสิทธิภาพของการส่งสัญญาณและการดำรงอยู่ของตัวรับสำรอง กลุ่มยา ปฏิกิริยาเหล่านี้แบ่งออกเป็นปฏิกิริยาต่อตัวรับและปฏิกิริยาทางเคมี
กลไกการเกิดปฏิกิริยาระหว่างยากับเครื่องปฏิกรณ์ชีวภาพ สามารถอธิบายได้ว่าเป็นโครงการต่อไปนี้: แต่ละแกนด์ (สารยาหรือสารตั้งต้นทางสรีรวิทยา) ผูกกับเว็บไซต์เฉพาะบนตัวรับเฉพาะ เปิดใช้งานเครื่องรับไอออนโดยตรงหรือโดยอ้อมควบคุมไอออนฟลักซ์ (1) และ / หรือกระบวนการภายในเซลล์อื่น ๆ (การหลั่งหรือการหดตัวของกล้ามเนื้อ) หรือเปิดใช้งานระบบโปรตีนนิวคลีโอไทด์ที่จับกับ Guanine (G-proteins) ซึ่งในทางกลับกัน ผู้ไกล่เกลี่ยที่แตกต่างกันหลายคนทำหน้าที่ในไซโตพลาสซึมกระตุ้นโปรตีนเป้าหมายต่าง ๆ เช่นไคเนสโปรตีน การกระทำหลังบนพื้นผิวที่เฉพาะเจาะจงและเป็นสื่อกลางในผลทางเภสัชวิทยา
จากคำอธิบายที่นำเสนอจะเห็นได้ว่าการกระทำของยาเสพติดดำเนินการโดยกลไกต่อไปนี้:
- ฟังก์ชั่นทางสรีรวิทยาของเนื้อเยื่อ (ตัวอย่างเช่น contractile, secretory) สามารถควบคุมได้โดยตัวรับหลายตัวและลิแกนด์ต่าง ๆ ;
- อาจมีหลายขั้นตอนกลางระหว่างการทำงานร่วมกันของสารยากับตัวรับและการตอบสนองของเนื้อเยื่อหรืออวัยวะโดยเฉพาะอย่างยิ่งการเปิดใช้งานของระบบที่เกี่ยวข้องกับผู้ไกล่เกลี่ยที่สองของผู้รับ;
- ประสิทธิผลของกลไกที่รับผิดชอบลำดับการตอบสนองต่อการกระตุ้นเช่นเดียวกับความหนาแน่นของตัวรับอาจแตกต่างกันไปตามเนื้อเยื่อ
ผลการรักษาของยาบางชนิดเกิดจากปฏิกิริยาทางเคมีโดยตรง (ไม่เกี่ยวข้องกับตัวรับเฉพาะ) กับสารประกอบภายนอกหรือกลไกอื่น ๆ ของการมีปฏิสัมพันธ์ (แรงดันออสโมติกการดูดซับ) ดังนั้นสำหรับยาขับปัสสาวะออสโมติก - แมนนิทอล, ยูเรีย - ไม่มีผู้รับเฉพาะ สารเหล่านี้เพิ่มแรงดันออสโมติกในท่อไตซึ่งเป็นผลมาจากการดูดซึมของน้ำถูกรบกวนและ diuresis เพิ่มขึ้น การกระทำของสารดูดซับ, ยาขับปัสสาวะสร้างกรดไม่เกี่ยวข้องกับตัวรับเฉพาะ
ยาลดกรด (เช่นอลูมิเนียมหรือแมกนีเซียมไฮดรอกไซด์) ทำปฏิกิริยากับกรดไฮโดรคลอริกเพื่อสร้างผลิตภัณฑ์ที่มีคุณสมบัติเป็นกรดอ่อน สารคีเลติ้ง, จับกับโลหะบางชนิดก่อให้เกิดสารประกอบเชิงเคมีที่ไม่ได้ใช้งาน
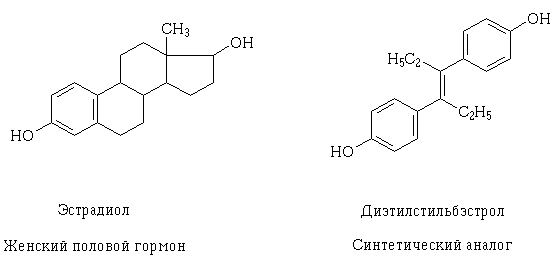
เมื่อความรู้เกี่ยวกับโครงสร้างของตัวรับและกลไกของปฏิกิริยาระหว่างเภสัชกรที่เป็นไปได้ของยาในระดับเซลล์ลึกลงไปการสร้างเป้าหมายของพวกเขาก็เป็นไปได้เช่นเดียวกับคำอธิบายว่าทำไมสารยาที่แตกต่างกันตั้งแต่แรก ตัวอย่างของปรากฏการณ์เช่นนี้คือ estradiol และ transisomer ของ diethylstilbestrol อะนาล็อกสังเคราะห์ของอวัยวะเพศหญิง โมเลกุลโครงสร้างของพวกมันแตกต่างกัน แต่มีกลุ่มไฮดรอกซีเดียวกันในคุณสมบัติและขนาดคล้ายกันและอยู่ในอวกาศเนื่องจากโมเลกุลของสารเหล่านี้สามารถโต้ตอบกับตัวรับเดียวกันและมีผลทางเภสัชวิทยาที่คล้ายกัน
วิธีการที่สารยาทำให้เกิดผลทางเภสัชวิทยาบางอย่างถูกใช้แทนคำว่า "กลไกการออกฤทธิ์" แนวคิดนี้ใช้เพื่ออธิบายผลกระทบของยาในระดับโมเลกุลอวัยวะและระบบ ตัวอย่างเช่นกลไกของการกระทำของตัวแทน anticholinesterase ที่ระดับโมเลกุลจะลดลงถึงการปิดล้อมของ acetylcholinesterase โดยการโต้ตอบกับศูนย์ประจุลบและ esterase ในขณะเดียวกันการอธิบายกลไกการเกิดฤทธิ์ลดความดันของยา anticholinesterase, bradycardia และ vasodilation นั้นเป็นสาเหตุของผลกระทบนี้นั่นคือพวกเขาพิจารณากลไกของผลกระทบนี้ในระดับอวัยวะ
การศึกษากลไกการออกฤทธิ์ของยายังคงดำเนินต่อไปและความคิดเกี่ยวกับกลไกการออกฤทธิ์ของสารเสพติดเนื่องจากข้อมูลใหม่ที่ได้รับไม่เพียง แต่จะมีรายละเอียดมากขึ้น แต่ยังเปลี่ยนแปลงอย่างมีนัยสำคัญ